Category: Uncategorized
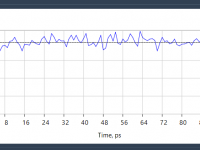
Why Your Molecular Simulations May Fail Without NVT Equilibration
Target Specific Atoms by Geometry in SAMSON: A Practical Guide for Molecular Modeling
One of the recurring challenges in molecular modeling is selecting atoms based on structural properties – particularly geometry. For instance, identifying atoms with tetrahedral geometry can be important for understanding chemical reactivity, assigning force field parameters, or preparing input for…
Make Models Fade Naturally with the Disappear Animation in SAMSON
From Molecules to Meshes: Exporting 3D Geometries with SAMSON
When You’re Stuck in Molecular Modeling, Try This
Effortless Scientific Storytelling: Using Background Animations in SAMSON Presentations
Making Molecular Highlights Stand Out with the Pulse Animation
Seeing Molecules Clearly: A Guide to Visual Models in SAMSON
Seeing Molecules: How Visual Models Make Molecular Modeling More Intuitive
Why Your Molecular Simulations May Fail Without NVT Equilibration
Target Specific Atoms by Geometry in SAMSON: A Practical Guide for Molecular Modeling
One of the recurring challenges in molecular modeling is selecting atoms based on structural properties – particularly geometry. For instance, identifying atoms with tetrahedral geometry can be important for understanding chemical reactivity, assigning force field parameters, or preparing input for…