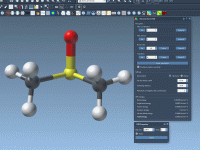
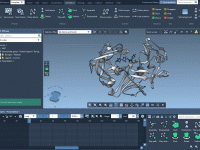
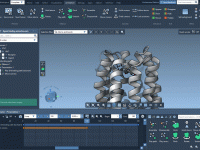
When working with force fields like the Universal Force Field (UFF) in molecular design, default automatic typing and bonding are often quite effective. However, when modeling molecules with unusual bonding patterns or working with partially known structures, default algorithms may…
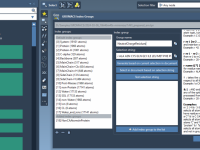
When setting up molecular simulations in GROMACS, creating custom index groups can be incredibly useful—for example, to define specific groups of atoms for pulling simulations or detailed analyses. But things tend to get tricky when working on large or batch…
When working with complex molecular systems, understanding and managing the size of your models is often essential. Whether you’re running simulations, visualizing, or preparing a publication, knowing the number of atoms—before diving deeper—can help you make decisions more efficiently and…
When preparing molecular animations or creating visual presentations, clarity is essential. Too many atoms or complex visualizations can make it difficult for a viewer to focus on the message a molecular modeler wants to convey. This is especially true for…
Creating compelling molecular animations can be challenging, especially when you’re trying to draw attention to specific regions of a complex system. A common frustration for molecular modelers is achieving smooth and expressive zooming effects that go beyond a simple magnification.…
Creating effective scientific animations often comes down to highlighting just the right parts of a molecular system at the right moment. If you’re preparing a presentation or a video to communicate your latest discovery, you probably don’t want all atoms…
As a molecular modeler working with DNA nanostructures, it’s common to design sophisticated geometries digitally, only to find yourself wondering how these can be simulated effectively. Bridging the gap between interactive design and coarse-grained simulation is essential — and this…
When starting out with molecular modeling in SAMSON, one of the first challenges you may encounter is how to make your molecular structures both informative and visually clear. Whether you’re preparing a figure for a publication or trying to make…
When working with large molecular systems, it’s common to end up with hundreds—or even thousands—of molecules in one document. Finding the exact molecule you’re interested in can feel like looking for a needle in a haystack. If you’re using SAMSON,…
When simulating molecular systems, it’s common to analyze dynamic regions that are central to your research – such as active sites, interface regions, or specific complexes. But as the system evolves over time, how can you always keep your eyes…