Category: Uncategorized
Saving Your Molecular Models: A Quick Guide to Exporters in SAMSON
When Hiding Is Better Than Fading: A Simple Trick in Molecular Animations
Solvating Coarse-Grained Systems in GROMACS Wizard: Avoiding Common Pitfalls
Building Repetitive Molecular Structures with SAMSON’s Pattern Editors
Designing nanoscale systems often requires repeated arrangements of atoms or molecular fragments—think helices, nanotubes, or crystal-like lattices. Yet, replicating and aligning these geometries manually can quickly become tedious and error-prone. That’s where SAMSON’s Pattern Editors come in: intuitive tools that…
Precise Molecular Alignment in SAMSON Without Scripting
Efficiently Setting Initial Conformations for Batch Simulations in SAMSON
When preparing molecular dynamics simulations, researchers often work with multiple starting conformations of the same system. This is especially common in studies involving ligand binding, protein flexibility, or comparative dynamics. However, managing and configuring these conformations for batch simulations can…
Quickly Locate and Manage Hidden Molecular Visual Models in SAMSON
Avoid Common Mistakes When Setting Up MARTINI Force Fields in Coarse-Grained Simulations
How to Select Atoms with Precision in SAMSON Using Mathematical Expressions
Saving Your Molecular Models: A Quick Guide to Exporters in SAMSON
When Hiding Is Better Than Fading: A Simple Trick in Molecular Animations
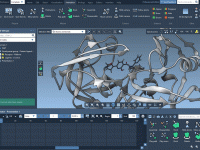
Solvating Coarse-Grained Systems in GROMACS Wizard: Avoiding Common Pitfalls
Building Repetitive Molecular Structures with SAMSON’s Pattern Editors
Designing nanoscale systems often requires repeated arrangements of atoms or molecular fragments—think helices, nanotubes, or crystal-like lattices. Yet, replicating and aligning these geometries manually can quickly become tedious and error-prone. That’s where SAMSON’s Pattern Editors come in: intuitive tools that…
Precise Molecular Alignment in SAMSON Without Scripting
Efficiently Setting Initial Conformations for Batch Simulations in SAMSON
When preparing molecular dynamics simulations, researchers often work with multiple starting conformations of the same system. This is especially common in studies involving ligand binding, protein flexibility, or comparative dynamics. However, managing and configuring these conformations for batch simulations can…