Category: Uncategorized
Making Molecular Models Clearer with the Right Colors
From Confusing to Clear: Managing Molecular Complexity with Visual Presets
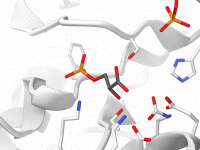
Visualizing molecular systems often feels like a balancing act—switching representations, adjusting colors, highlighting key atoms—before you’ve even started analyzing. For researchers and students dealing with increasingly large biomolecular structures, the time spent configuring these visualizations manually can quickly add up.…
Creating a Pause for Emphasis in Molecular Animations
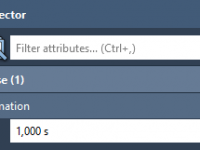
When preparing molecular presentations, one common challenge researchers face is making specific moments stand out. Whether you’re highlighting a protein-ligand interaction or illustrating a crucial conformational change, these scientific storytelling moments need time to register with your audience. Animations move…
A Simpler Way to Hide Atoms in Molecular Animations
When creating molecular animations, many researchers and educators face a common challenge: controlling the visibility of individual atoms without relying on transparency settings, which can lead to cluttered visuals and overlapping structures. Whether you’re preparing a molecular dynamics presentation or…
When Reproducibility Matters: Running GROMACS Simulations with Your Preferred Version
Keeping Track of Cloud Simulations with SAMSON’s Job Manager
What If You Could Just Talk to Your Molecular Modeling Platform?
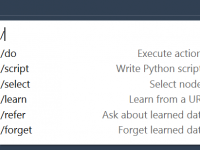
Many molecular modelers spend significant time navigating menus, remembering shortcuts, and writing scripts to perform everyday tasks: visualizing surfaces, modifying representations, or selecting atom groups. While flexibility is appreciated, the friction can add up—especially when switching between modeling tools and…
When It Matters: How to Use Your Own Version of GROMACS in SAMSON
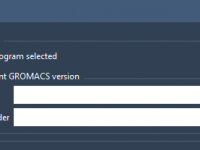
Switching between different versions of GROMACS isn’t just a technical preference—it’s often a necessity for molecular modelers. Whether for ensuring reproducibility, taking advantage of specific performance improvements, or simply using a cluster’s precompiled GROMACS, being able to control the version…
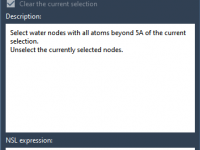
Saving Key Waters: Selecting and Deleting Crystal Waters Outside the Active Site
Making Molecular Models Clearer with the Right Colors
From Confusing to Clear: Managing Molecular Complexity with Visual Presets
Visualizing molecular systems often feels like a balancing act—switching representations, adjusting colors, highlighting key atoms—before you’ve even started analyzing. For researchers and students dealing with increasingly large biomolecular structures, the time spent configuring these visualizations manually can quickly add up.…
Creating a Pause for Emphasis in Molecular Animations
When preparing molecular presentations, one common challenge researchers face is making specific moments stand out. Whether you’re highlighting a protein-ligand interaction or illustrating a crucial conformational change, these scientific storytelling moments need time to register with your audience. Animations move…
A Simpler Way to Hide Atoms in Molecular Animations
When creating molecular animations, many researchers and educators face a common challenge: controlling the visibility of individual atoms without relying on transparency settings, which can lead to cluttered visuals and overlapping structures. Whether you’re preparing a molecular dynamics presentation or…
When Reproducibility Matters: Running GROMACS Simulations with Your Preferred Version
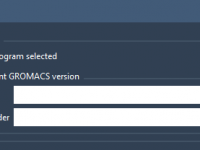
Keeping Track of Cloud Simulations with SAMSON’s Job Manager
What If You Could Just Talk to Your Molecular Modeling Platform?
Many molecular modelers spend significant time navigating menus, remembering shortcuts, and writing scripts to perform everyday tasks: visualizing surfaces, modifying representations, or selecting atom groups. While flexibility is appreciated, the friction can add up—especially when switching between modeling tools and…
When It Matters: How to Use Your Own Version of GROMACS in SAMSON
Switching between different versions of GROMACS isn’t just a technical preference—it’s often a necessity for molecular modelers. Whether for ensuring reproducibility, taking advantage of specific performance improvements, or simply using a cluster’s precompiled GROMACS, being able to control the version…