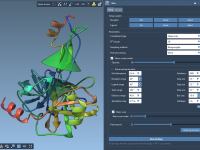
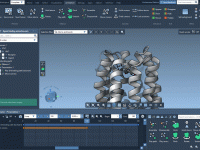
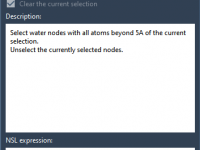
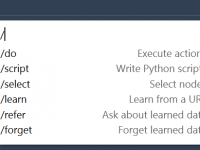
When working with complex molecular systems, it’s not unusual to feel overwhelmed by the sheer amount of structural data at your fingertips. Whether you’re preparing a model for visualization, simulation, or export, quickly narrowing down to specific features can save…
One of the most common challenges molecular modelers face during protein-protein docking is handling the vast number of potential orientations and false positives generated during the search. Even when docking crystal structures, an unrestricted search can lead to misleading predictions…
Do you ever find yourself losing sight of key atoms when playing or exporting molecular animations? When tracking dynamic molecular systems, it’s common to want the camera to remain stationary while directing your attention (and the viewer’s) toward something changing…
One of the subtle but essential tasks in preparing a molecular system for a GROMACS simulation is deciding which water molecules to keep. While removing all crystal water molecules might simplify pre-processing, it may also mean losing functionally relevant water…
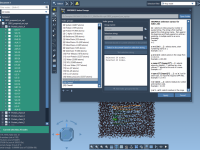
For many molecular modelers, writing Python scripts is a common task—but it can also be a source of friction. Whether you’re building custom visualizations, automating repetitive tasks, or analyzing structural properties, scripting can be both powerful and time-consuming. But what…
If you’ve ever tried to simulate a system with multiple replicas of the same protein and ran into topology generation errors, you’re not alone. This is a common issue when working with coarse-grained modeling tools like Martinize2 in SAMSON. The…
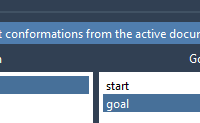
Many molecular modelers face a recurring challenge when studying functional protein dynamics: how to connect two known conformations of a protein—say, an open and a closed state—with a realistic transition path. This is a critical step for gaining insights into…
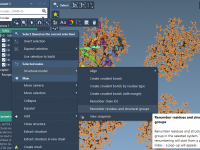
When setting up a simulation involving custom pulling forces in GROMACS, one of the first roadblocks many molecular modelers face is the creation of appropriate index groups. These groups are essential to define regions of interest in biomolecular simulations—whether you’re…
When working on complex molecular systems in SAMSON, we often deal with a multitude of objects: atoms, groups, models, data nodes, and more. Among these, property models define additional data on molecular systems, such as electrostatic potentials, surface properties, or…
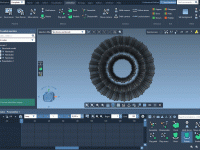
Adding motion to molecular simulations can help communicate structural dynamics and emphasize specific interactions. One common need in molecular modeling is showcasing how a group of atoms or residues rotates in 3D space—whether for presenting conformational changes or emphasizing the…