





When modeling conformational transitions between protein states, one of the most overlooked yet crucial steps is structure preparation. Whether you’re setting up umbrella sampling, visualizing intermediate states, or refining with steered molecular dynamics, how you prepare your structures can determine whether your workflow runs smoothly or ends up halted by cryptic errors.
In this article, we focus on a common issue in conformational pathway modeling: mismatch and disconnected components when interpolating between structures. We’ll walk through how to properly prepare protein structures for interpolation using the As-Rigid-As-Possible (ARAP) Interpolator in SAMSON.
Why preparation matters
Suppose you’re interpolating between two conformations of a protein using ARAP. You get everything ready, hit run, and suddenly an error appears:
“Cannot proceed because the structure does not make one connected component.”
This often happens because the input structures still include water molecules, ligands, ions, or alternate locations. These elements can introduce disconnected atoms, breaking the algorithm’s assumption of structural continuity.
Quick start: Cleaning your structures
Still using unprocessed raw PDB files? Here’s how to clean them inside SAMSON:
- Load the structures you’d like to interpolate using Home > Fetch. For example, try loading
1DDTand1MDTfrom the Protein Data Bank. - Open each structure in the Document view and remove any chains you won’t use. In this tutorial, we focus only on chain
A. - Use the Home > Prepare function. This automatically removes water, ligands, ions, and alternate locations.
Below is a snapshot from the documentation showing the process of removing chain B from one of the input structures:
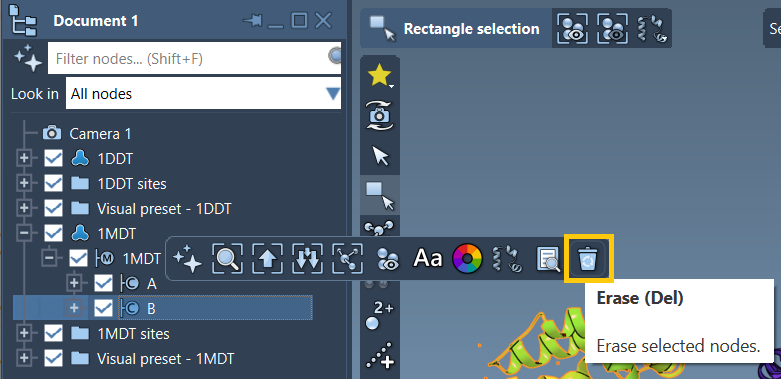
Once cleaned, the structures will behave more predictably and agree structurally, making the interpolation between conformations smoother and more reliable.
Common pitfalls and how to avoid them
- Retain only the relevant chain: For multi-chain PDBs, extra chains can lead to atom mismatching. Limit your structures to only the chains of interest.
- Check for alternate positions: Alternate side chain conformations can confuse atomic matching. Make sure only one position per atom is retained.
- Confirm connectivity: Visual inspection in SAMSON can help. In the Document view, disconnected pieces are usually visible. Delete unnecessary fragments.
Still receiving errors?
If you’ve run Home > Prepare and still get connectivity errors, ensure the entire structure forms a single connected component. Any floating ions, small molecules, or solvent molecules need to be removed manually.
Conclusion
Proper protein preparation is essential before running ARAP interpolation or any workflow requiring atom matching. It helps avoid hard-to-debug errors and ensures that what you’re analyzing reflects the motions of your protein — not artifacts from uncleaned data.
To learn more and walk through a full example, visit the full tutorial at:
https://documentation.samson-connect.net/tutorials/arap/arap-interpolation-for-protein-structures/
SAMSON and all SAMSON Extensions are free for non-commercial use. To get started, download SAMSON at https://www.samson-connect.net.