





Finding saddle points and minimum energy paths (MEPs) between molecular conformations can be computationally expensive. The Parallel Nudged Elastic Band (P-NEB) method in SAMSON is designed to optimize such transitions by refining intermediate images along a proposed path. But depending on how you set up your input, you might save significant time during optimization ⏱️.
One common question for molecular designers new to P-NEB workflows is: Should I use conformations or paths as input? The answer: use paths whenever possible.
Why it matters: performance and efficiency
The P-NEB algorithm can be applied to either:
- a path — a trajectory object that defines how a group of atoms move over time
- a set of conformations — independent snapshots of atom positions
While both configurations are supported, applying P-NEB to a set of conformations requires a separate optimization thread for each step in the set. This leads to longer computation times — especially for complex systems with many atoms or many intermediate states.
🧪 Tip: If you already have conformations, you can combine them into a path by selecting them in the Document view and right-clicking:
Conformation > Create path from conformations.
What this looks like in SAMSON
Let’s walk through how P-NEB optimization works in both cases. First, from the Document view, either select a path or a set of conformations.
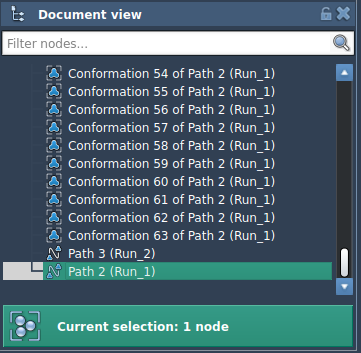
Then, open the P-NEB app from Home > Apps > All > P-NEB and click Run. Choose the following setup:
- Spring constant: 1.00
- Number of loops: 100
- Interaction model: Universal Force Field
- Optimizer: FIRE
- Parallel execution: ✔️ checked
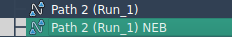
When applied to a path, P-NEB produces a new, optimized trajectory with uniform spacing and minimized energy gradients:
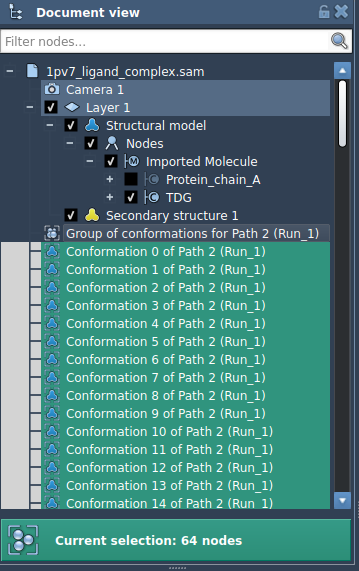
If instead you optimize a set of conformations:
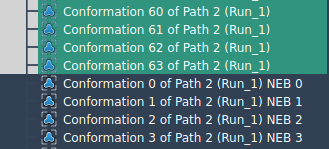
… the computation will run, but expect longer times:
Both workflows result in valuable data on molecular transitions. But from a practical perspective — especially when exploring many candidate paths — choosing to work with paths over raw conformations can lead to major time savings, smoother user experience, and easier visualization.
Curious to try this in your own workflow? You can explore the full tutorial and sample systems at the original documentation page: Optimize Transition Paths with the Parallel Nudged Elastic Band method.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here.