





Protein structures can adopt multiple conformations, critical for understanding their function in biological systems. Modeling a smooth, realistic transition path between two such conformations can be a challenge for molecular modelers, especially under time constraints or when aiming for high accuracy. Fortunately, the As-Rigid-As-Possible (ARAP) Interpolator in SAMSON provides a solution that is both efficient and detailed. In this blog post, we’ll guide you through generating a seamless transition path between two protein conformations in just a few steps.
Why Model Conformational Transitions?
Visualizing conformational transitions is essential for multiple applications, such as:
- Understanding protein dynamics and functional states.
- Setting up free energy simulations with realistic reaction coordinates.
- Preparing inputs for advanced computational techniques like umbrella sampling or nudged elastic band methods.
If you’ve ever struggled with aligning conformations or tackling computationally expensive methods for interpolation, the ARAP Interpolator addresses these pain points with a user-friendly and fast approach.
Step-by-Step Guide to Using the ARAP Interpolator
1. Prepare Your Protein Structures
Start by loading the structures of interest directly in SAMSON. For example, you might use two conformations of the Diphtheria Toxin (PDB IDs: 1DDT and 1MDT):
- Open Home > Fetch and input
1DDT 1MDT. - Load the structures and keep only chain
Aof each. - Run Home > Prepare to clean the structures (remove waters, ligands, etc.).
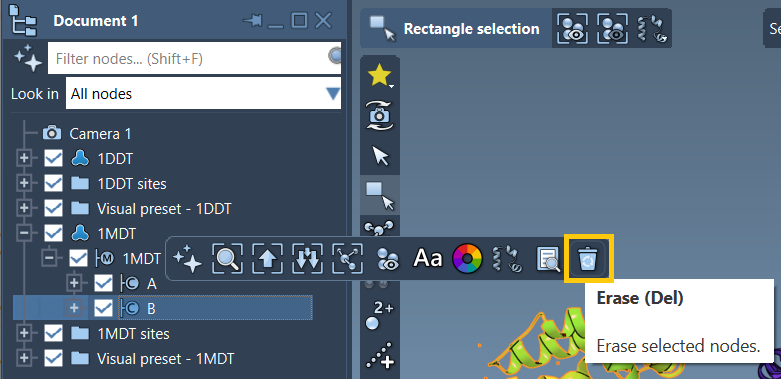
2. Define Conformations
Select the cleaned structures and define conformations for each:
- For
1DDT, click Edit > Conformation and name it1DDT A. - Repeat for
1MDTand name its conformation1MDT A.
These conformations act as the start and goal points of your transition path.
3. Run the ARAP Interpolation App
To compute the transition:
- Open Home > Apps > Biology > ARAP Path Interpolation.
- Choose Start:
1DDT Aand Goal:1MDT A. - Match atoms using All except hydrogens for precision.
- Configure edge construction by selecting from bonds in the Start structure and Try connecting α-carbons before and after missing residue segments.
Set the number of path conformations (e.g., 20) and click Run. In seconds, your path is generated!
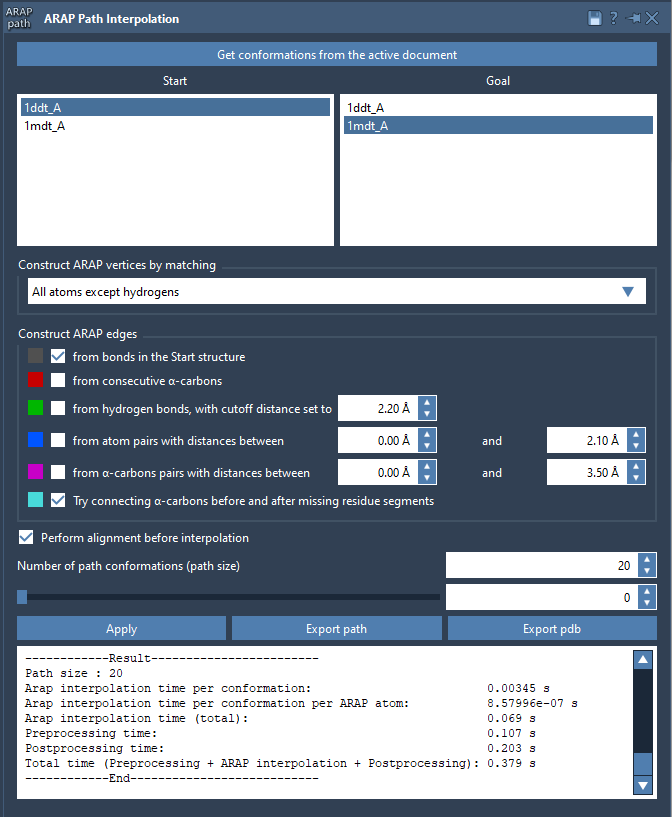
4. Visualize and Export
Once the interpolation is complete, use the app slider to explore intermediate conformations in the path:
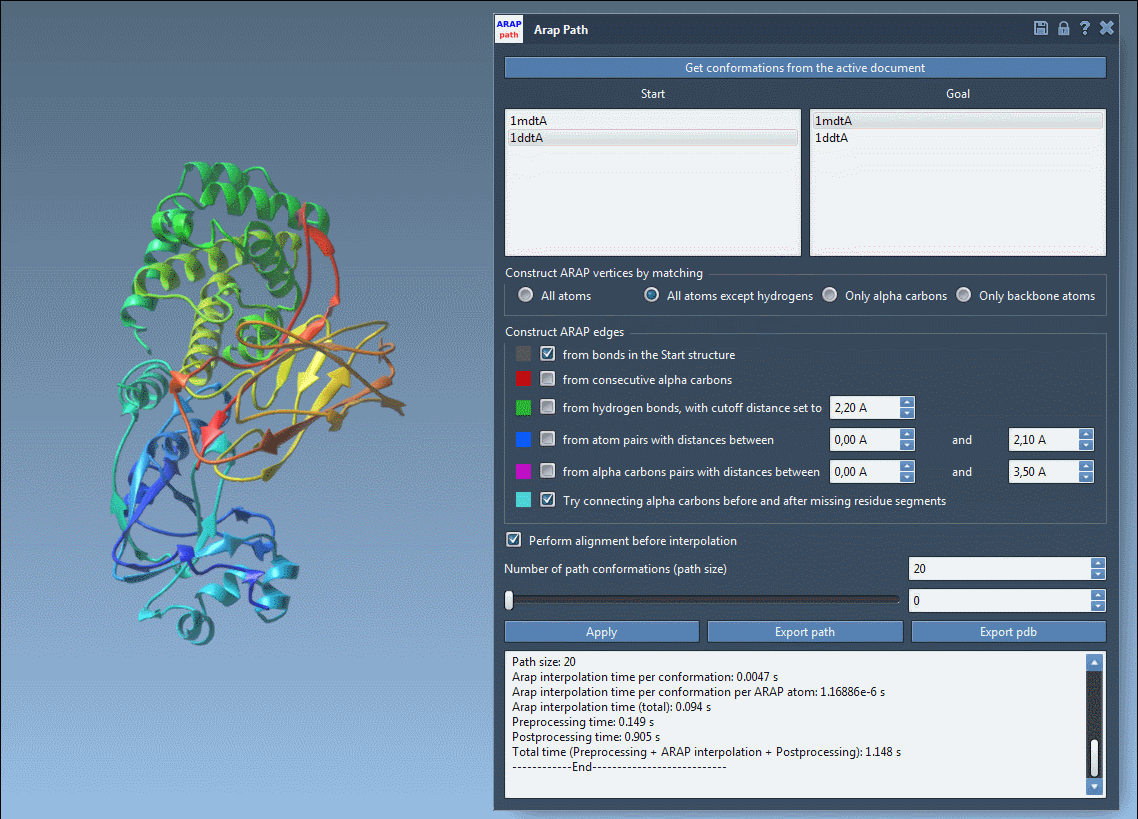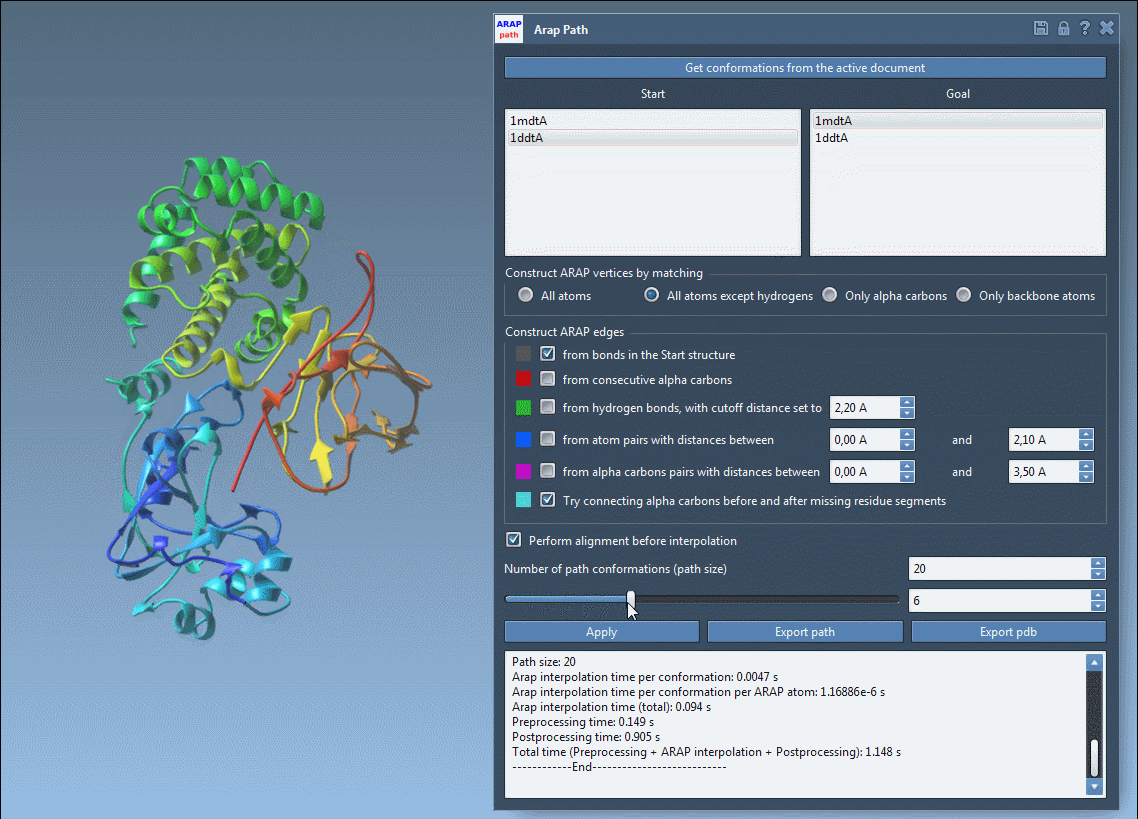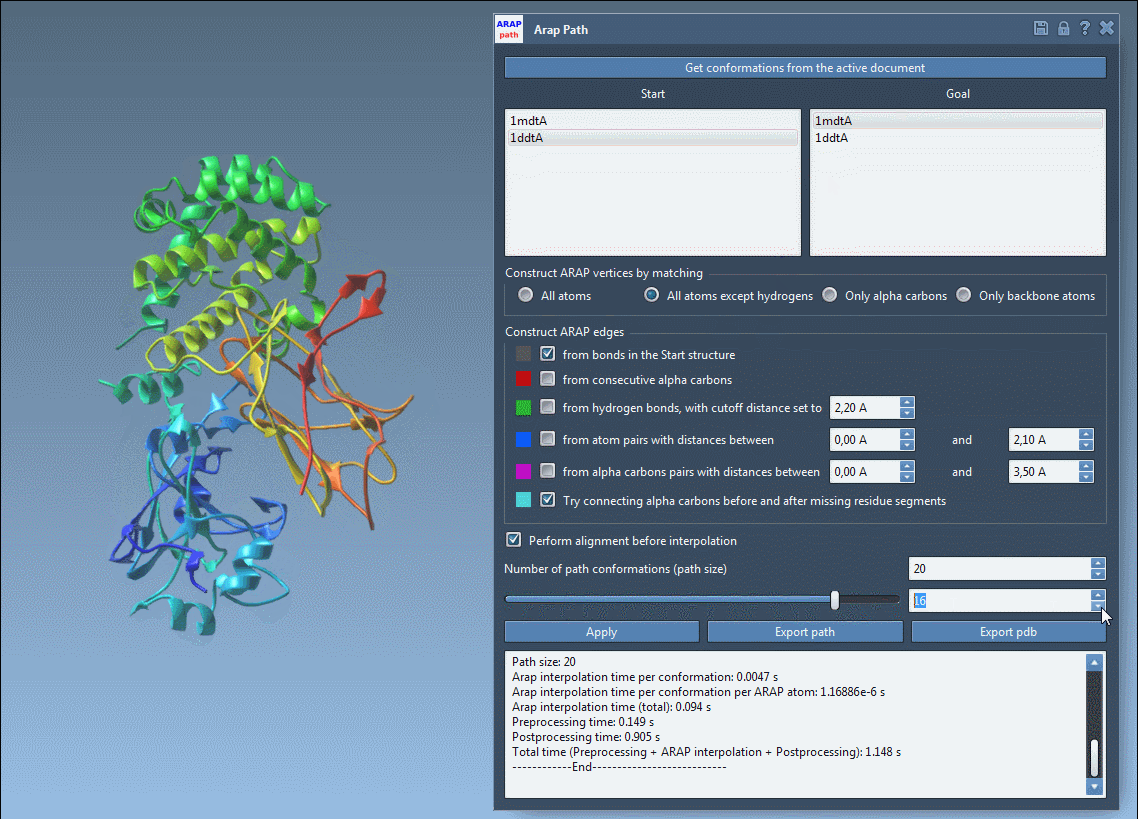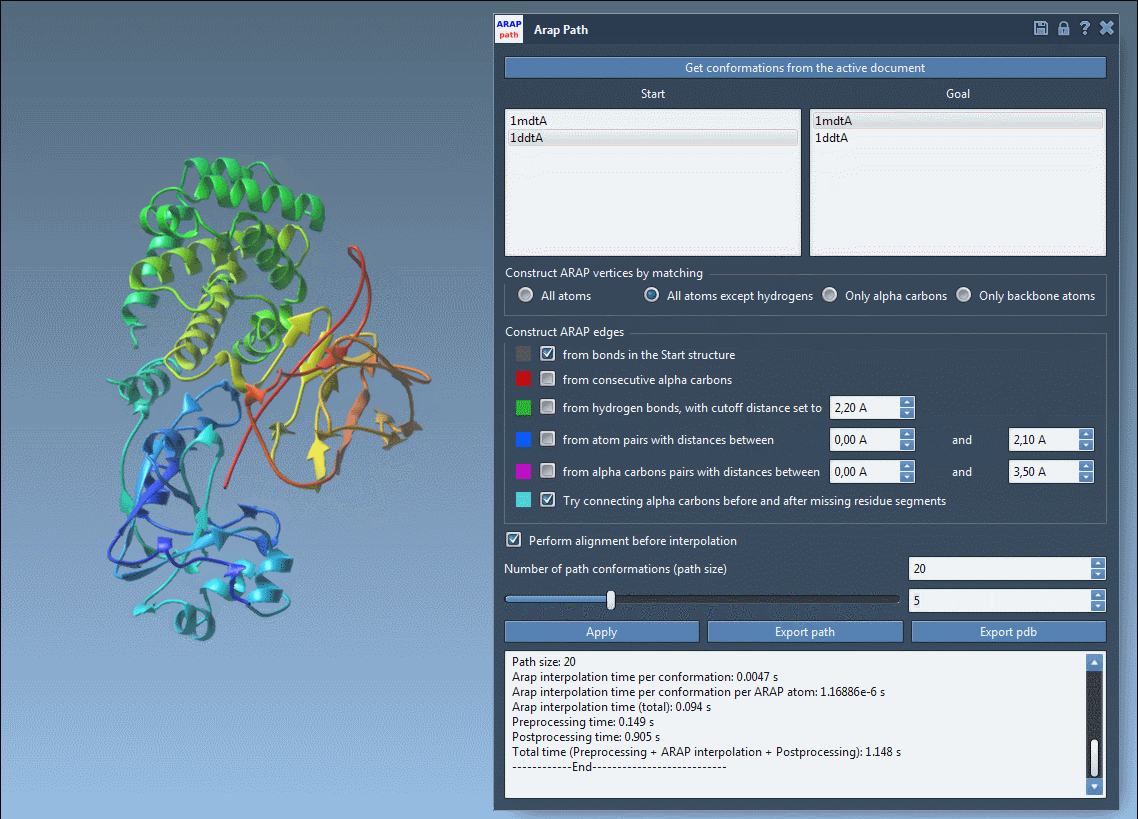
You can export the results either as a trajectory within SAMSON or as PDB files for further analysis in other tools.
A Key Feature for Molecular Modelers
Whether you’re preparing inputs for molecular simulations or simply analyzing structure-to-structure transitions, the ARAP Interpolator simplifies a complex task into a series of intuitive steps. Save time and refine your workflows with this powerful tool.
To dive deeper into the details, visit the full documentation at ARAP Interpolation for Protein Structures.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON at SAMSON Connect.