





Setting up a molecular dynamics simulation that involves a reaction coordinate—such as a ligand unbinding pathway—can be time-consuming. One frequently reported pain by molecular modelers is the extraction of atomic coordinates along a pre-defined path for use in free energy profiling. Manually scripting coordinate extraction often leads to errors or inefficiencies.
If this sounds familiar, the Export Along Paths Extension in SAMSON may help streamline your workflow. This powerful yet intuitive app simplifies the process of exporting atomic trajectories along paths—whether for all atoms or just a subset like a ligand. It’s especially useful after you’ve identified a pathway using tools like Ligand Path Finder and optimized it using techniques such as the nudged elastic band (NEB) method.
What’s the Use Case?
If you’re preparing input data for:
- Umbrella sampling
- String method simulations
- Path collective variable setups
…then exporting coordinates at intervals along a molecular pathway becomes necessary. The Export Along Paths Extension makes this process interactive and visual, with no need for custom scripts.
Focus: Exporting a Subset of Atoms (e.g., Ligands)
Here’s a quick walkthrough that shows how to export a subset of atoms—say, just the ligand—along an unbinding path. We’ll use the sample system included in the tutorial: Thiodigalactosid (TDG) unbinding from Lactose permease (PDB ID 1PV7).
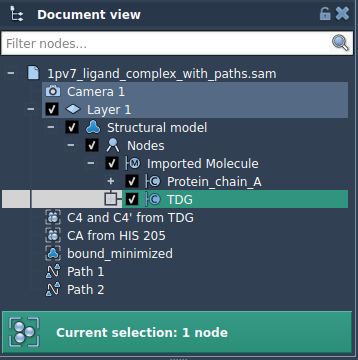
- Select the subset you’re interested in, such as the TDG ligand, in the Document view.
- Open the Export Along Paths app via Home > Apps > All or by typing Shift+E.
- From the Advanced panel, click Add to store your selected atoms as a model for export.
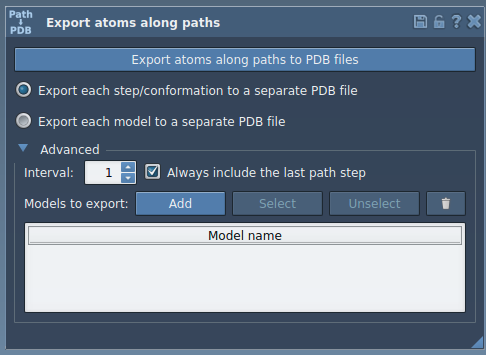
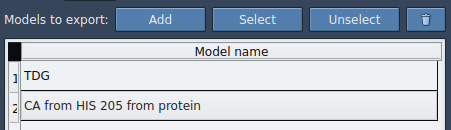
Once added, your selection appears in a table of named models. You can:
- Rename each model
- Include multiple groups of atoms (e.g., ligand and binding site)
- Reset or reselect model definitions
To export:
- Choose whether you want a single PDB file for all frames, or one per frame.
- Select the relevant path(s) in the Document view.
- Click Export atoms along paths to PDB files.
You’ll then be prompted to choose a destination folder and file prefix. SAMSON takes care of the rest—frames are exported based on the path, and only for the atoms you care about.
This is particularly useful when preparing starting points for simulations that require atomic coordinates at different stages along a path. For example, you might extract the ligand’s positions from a 40-frame path to set up windows for umbrella sampling.
Try It With a Sample System
A ready-to-use molecular model, complete with a protein-ligand complex and precomputed paths, is available in the documentation. This makes it easy to reproduce and understand each step before applying the same logic to your own system.
Learn more by visiting the full tutorial at the SAMSON documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.