





For molecular modelers, achieving accurate geometry refinement quickly can be a challenging task. Whether you’re exploring molecular structures, refining inorganic materials, or simulating catalyst systems, the UMA Force Field in SAMSON provides a machine-learning-based solution designed for high responsiveness and adaptability. This blog post explores the key steps to utilize UMA Force Field for rapid and insightful geometry refinement, helping you optimize your workflows more effectively.
Why use UMA Force Field for geometry refinement?
The UMA Force Field is tailored to handle diverse atomistic systems with robust and responsive performance. It supports interactive energy and force evaluation, enabling users to refine geometries, explore structural properties, and gain insights efficiently. By leveraging pre-trained machine-learning models, UMA offers three model variants depending on your specific computational needs:
- UMA-S (2.3 GB, v1.2): Ideal for rapid workflows and interactive tasks.
- UMA-S (1.1 GB, v1.1): A lightweight legacy option for less demanding systems.
- UMA-M (11.7 GB, v1.1): Provides enhanced capacity for complex systems.
Choosing the optimal model allows for a balance between speed and computational demand based on your needs.
Setting up UMA for geometry refinement
Getting started with UMA Force Field involves a few straightforward steps:
- Open a document containing the molecular or material system you want to refine within SAMSON.
- Navigate to
Edit > Simulate > Add simulatorand select the UMA Force Field interaction model from the list. - Choose a state updater for your simulation, such as FIRE (Fast Inertial Relaxation Engine).
- Click OK to confirm your selections and proceed.
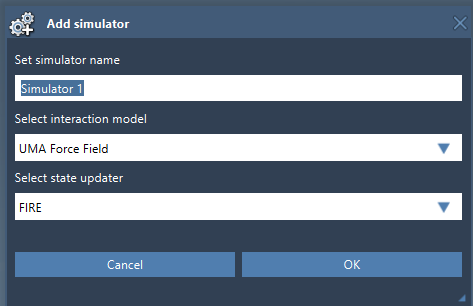
Following this, the UMA Force Field Setup window will appear. You will need to specify the Model and Task. The flexibility of UMA is evident in its diverse application domains, such as:
- OMOL – Molecules: For molecular systems using charge and spin multiplicity.
- OMAT – Inorganic materials: Optimized for bulk inorganic systems.
- ODAC – MOFs: Molecular dynamics for Metal-Organic Frameworks and adsorption workflows.
- OC20 – Catalysis: For catalytic surfaces and adsorbates.
- OMC – Molecular crystals: Tailored for molecular crystal systems.
Running your UMA simulation
Once set up, you can begin the simulation by selecting Edit > Simulate > Start. UMA initializes its backend environment, loads the selected model, and opens the UMA Properties window. From here, you can visualize simulation properties such as energy updates and interactively refine your system by modifying atom positions. At this stage, UMA dynamically updates energy and force calculations, providing real-time feedback.
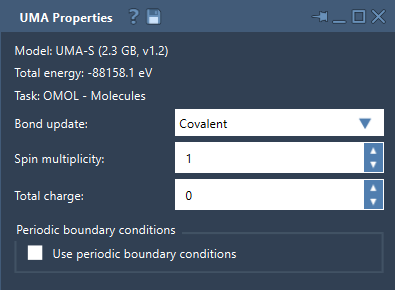
If you need to fine-tune specific aspects, such as bond graphs or spin states, UMA’s interface allows adjustments via:
- Bond Update Mode: Options for recomputing covalent bonds, estimating Wiberg or Mayer bond orders, or maintaining the current bond graph unchanged.
- Charge and Spin Multiplicity: Useful for molecular tasks (OMOL) to define system-specific electronic properties.
- Periodic Boundary Conditions: Enable periodic simulations with a 3 x 3 cell matrix configuration.
Best practices for refinement workflows
To ensure efficient use of the UMA Force Field, consider the following tips:
- Select a task that closely aligns with your system type.
- Start with UMA-S for faster iterations and only switch to UMA-M if additional capacity is needed.
- For molecular systems, ensure total charge and spin multiplicity align with the intended system properties.
- Verify periodic unit cells before interpreting results for periodic workflows.
- Use UMA for exploratory and structural refinement, but finalize with a high-accuracy reference method when required.
To learn more about refining geometries and optimizing workflows with UMA, visit the complete UMA Force Field documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get your copy at https://www.samson-connect.net.