





Protein structure alignment is a critical step for molecular modelers, particularly when trying to understand conserved regions, functional residues, or structural differences between homologous proteins across species. Yet, for many, this process can feel daunting and time-consuming. Fortunately, SAMSON’s Protein Aligner extension simplifies this workflow, providing a seamless tool to align protein sequences and superimpose their structures efficiently.
In this post, we’ll walk you through performing protein structure alignment with SAMSON and highlight features that will make this process not only easier but also more efficient.
Why is Protein Alignment Useful?
Protein alignment isn’t just about structural superimposition—it allows researchers to:
- Identify conserved residues that facilitate function or ligand binding, which are crucial for protein engineering or drug development.
- Compare protein conformations across species or mutated variants.
- Assist in generating accurate homology models for downstream molecular design workflows.
Yet, performing these tasks manually often involves juggling various tools and formats. That’s where SAMSON’s intuitive features stand out.
Getting Started with Protein Aligner
To begin, ensure you have SAMSON installed, along with access to Protein Aligner. Start by loading the proteins you want to align. For this example, we’ll superimpose two hemoglobins from different organisms (1DLW and 1RTX).
- In SAMSON, navigate to Home > Fetch.
- Input the PDB identifiers
1DLW 1RTXin the fetch dialog. Choose either PDB or PDB (mmCIF), then click Load.

If the structures you load contain extra solvent or ligands, clean them up first using Protein Preparation & Validation, accessible via Home > Prepare.
Launching Protein Aligner
After your proteins are loaded, open the Protein Aligner from Home > Align. This brings up an interface that lets you align sequences, highlight conserved regions, and superimpose structures efficiently.

Whole-Protein Superposition
Superimposing entire proteins is straightforward:
- Ensure no residues are selected beforehand.
- Click Align to this on the first protein model’s row.
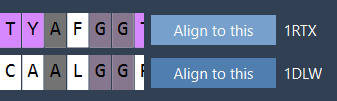
After alignment, the RMSD (Root Mean Square Deviation), an important metric for evaluating structure similarity, will appear between the aligned structures. For example, you might see a reported RMSD of 3.27 Å:

Your proteins are now superimposed. You can adjust visualization settings, like adding secondary structure ribbons, to better understand the alignment. Simply navigate to Visualization > Visual model > Ribbons, and consider coloring each ribbon differently for clarity.
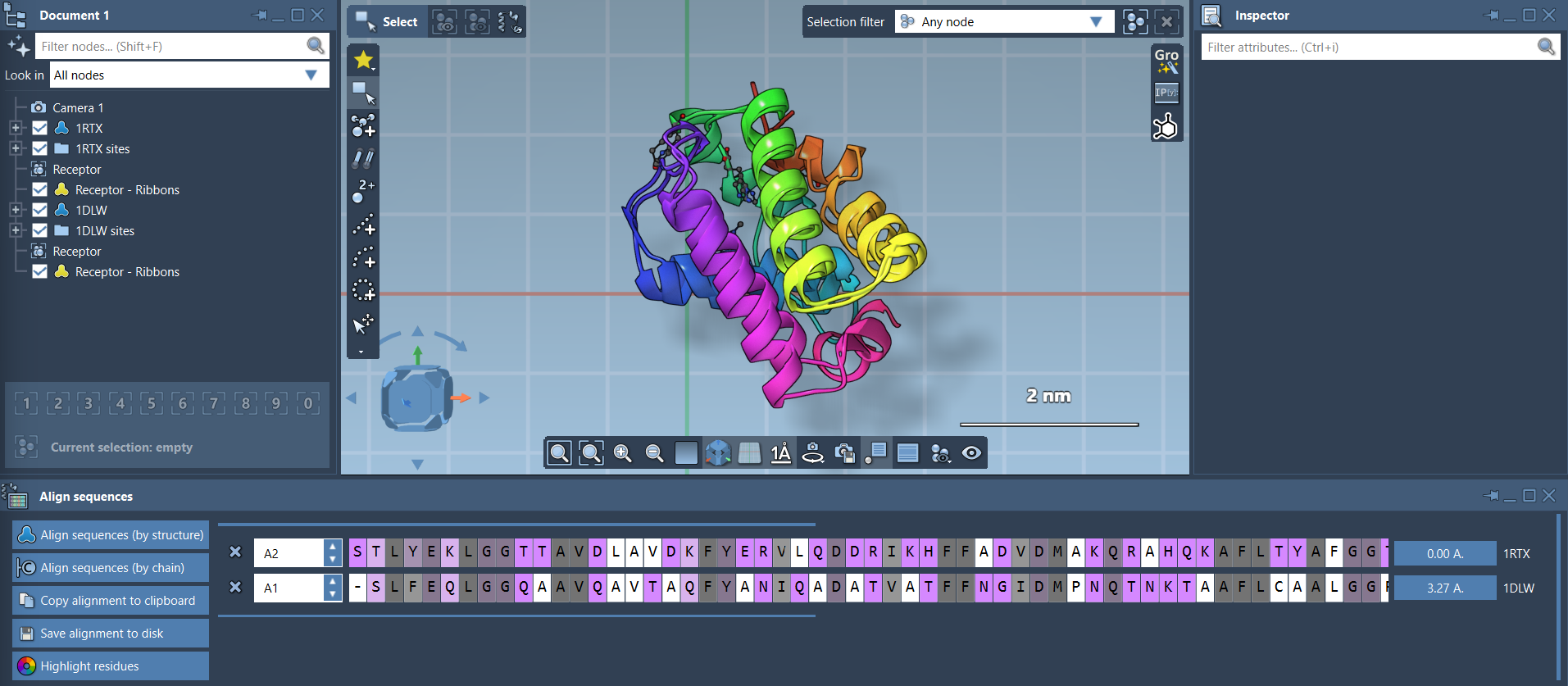
Refining Your Alignment to Specific Regions
What if you’re only interested in a specific region, like an alpha-helix or a binding pocket? SAMSON makes this easy as well. For example, if you’re focusing on the first 20 residues of these hemoglobins:
- Select the first 20 residues in both sequences directly in the Protein Aligner interface.
- Click the alignment button (e.g., the
0.0 Åbutton) next to this selection to superimpose only the targeted residues.

The result is a precise superimposition of the chosen region without unwanted influence from other parts of the protein structure:

What’s Next?
After aligning your proteins, you can:
- Export the alignment for homology modeling.
- Analyze conserved residues in ligand-binding sites.
- Repeat the alignment process with additional chains or related proteins.
Want to explore Protein Aligner further? Visit the official documentation to learn more about how it can enhance your workflows.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at samson-connect.net.