





One of the most important challenges in molecular modeling today is understanding the dynamic processes of critical biomolecules. The SARS-CoV-2 spike protein is particularly intriguing due to its role in binding to human cells and facilitating viral entry. If you’re trying to model the motion of this protein as it transitions from its closed to open state, the following guide will give you an efficient pathway to compute this motion directly using SAMSON, the integrative molecular design platform.
Why model spike motion?
The SARS-CoV-2 spike protein is central to the virus’s infectivity. Its ability to bind to the human ACE2 receptor and facilitate cell entry is a dynamic process, requiring an open spike conformation. Understanding how this motion unfolds can aid in vaccine and drug design efforts by providing insights into potential therapeutic targets. Modeling this motion effectively often requires addressing challenges like discrepancies in structural data or incorporating smooth transitions between intermediate states.
How SAMSON solves this problem
SAMSON provides tools that can simplify the process of computing the spike protein motion from a closed to an open state. By integrating methods like ARAP Interpolation Path and Parallel Nudged Elastic Band (P-NEB), SAMSON allows researchers to calculate motion efficiently while incorporating structural details from cryo-EM and X-ray crystal structures. Here’s a step-by-step guide on how this was done for the SARS-CoV-2 spike protein:
- Start with the known structures: Researchers used the closed conformation (PDB 6VXX) and the open conformation (PDB 6VYB) of the spike protein as input. These structures provide the foundation for interpolating the motion.
- Prepare the data: Both structures were pre-processed by modifying bond orders in sugar residues using a Python script. Hydrogens were added and the structures were minimized to ensure compatibility for further calculations.
- Generate an initial path: The ARAP Interpolation Path module was used to compute an interpolated motion path from the open state to the closed state. This step only took under a minute on a standard laptop, demonstrating SAMSON’s efficiency.
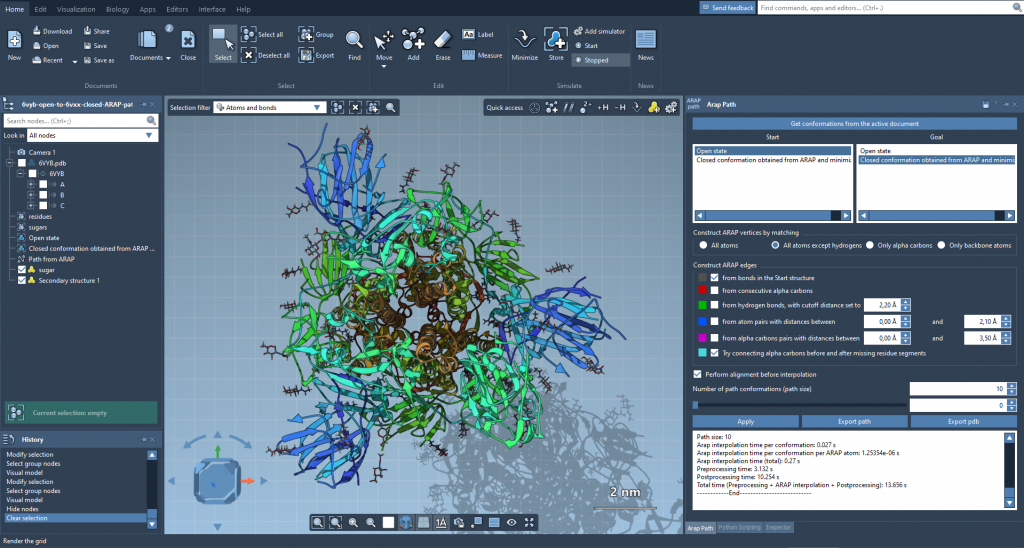
- Refine the model: Due to the difference in residues between the two conformations, intermediate closed-state conformations were extracted from the ARAP results and further minimized. This helped align the intermediate states to the open conformation structure.
- Improve with P-NEB: The P-NEB module was used to refine the interpolated path by focusing on energy minimization. The refinement step took only about 15 minutes, even for a computationally intensive process.
The results
The final result provides a detailed trajectory of how the spike protein transitions from its closed state to the open state. This motion can be visualized in different angles, and the data is available for download in several formats, including PDB, SAMSON format, and trajectory files. The motion showcases how one of the spike proteins in the trimer opens to bind the ACE2 receptor, a critical step in viral infection.
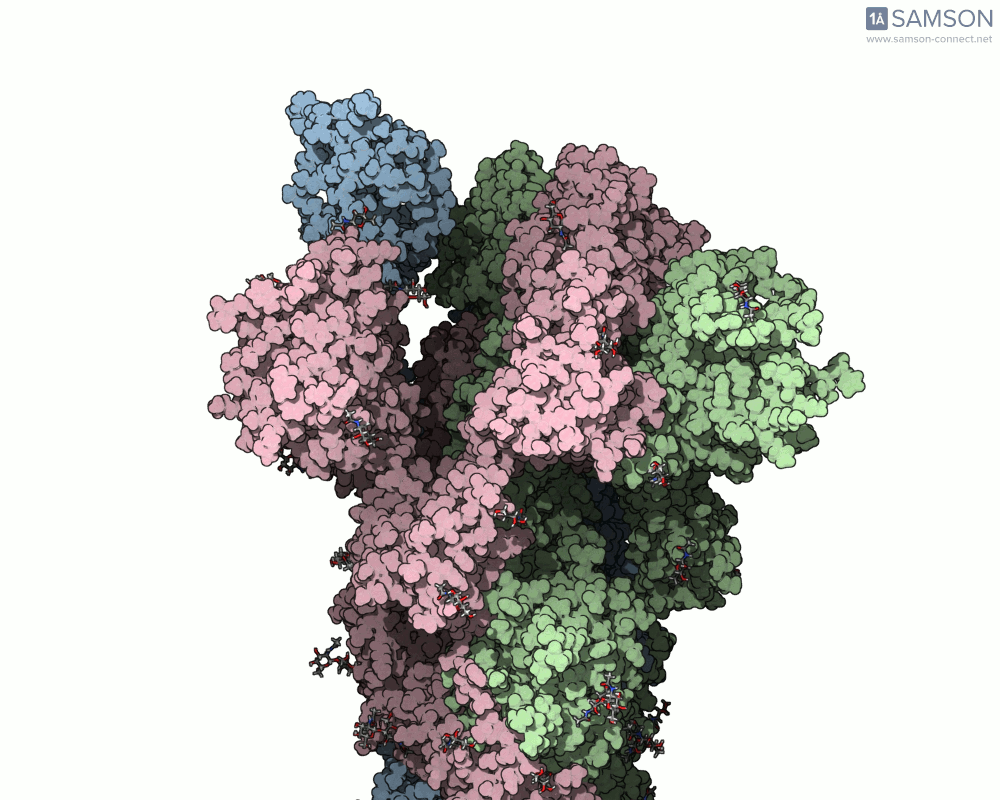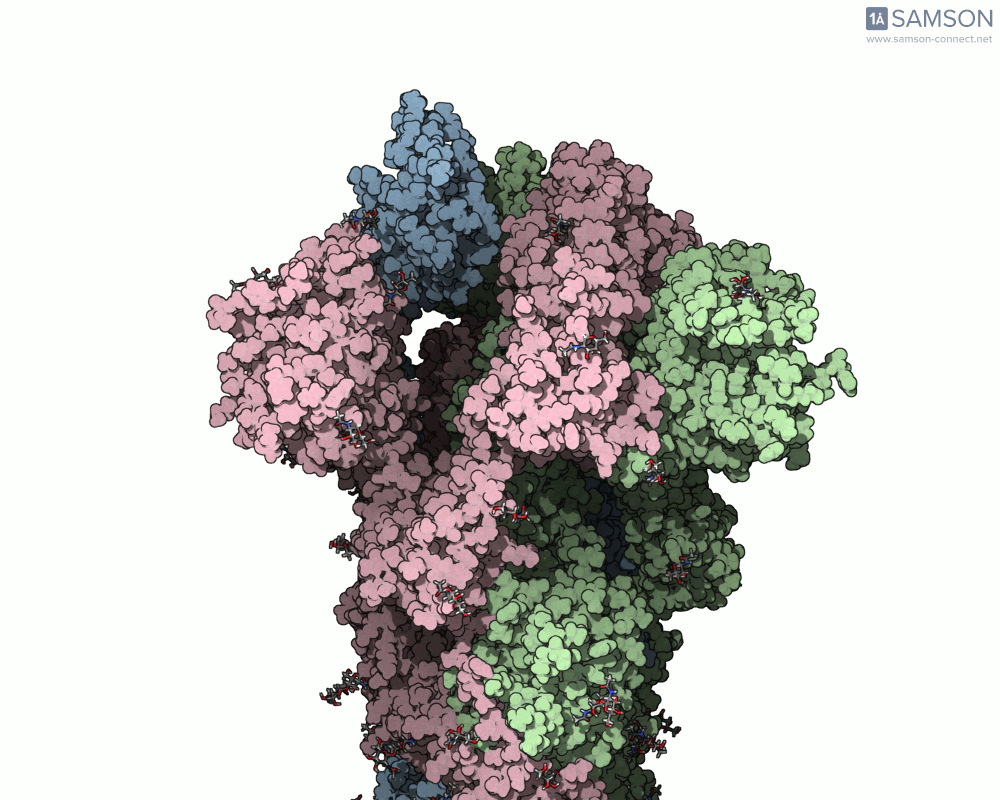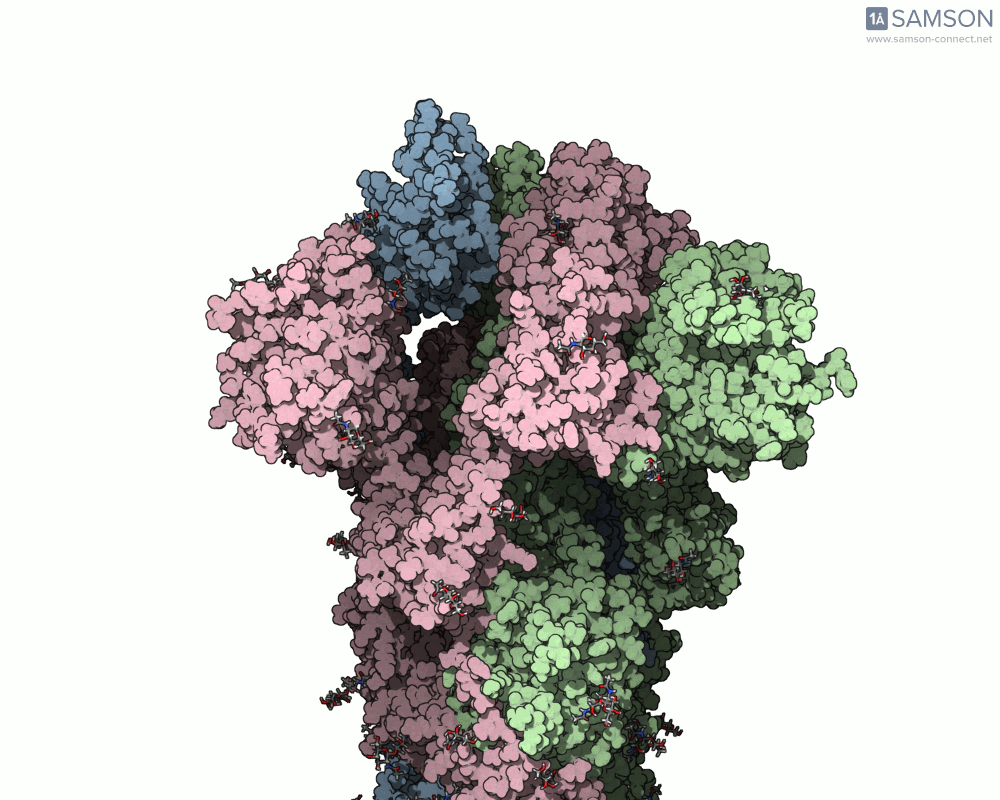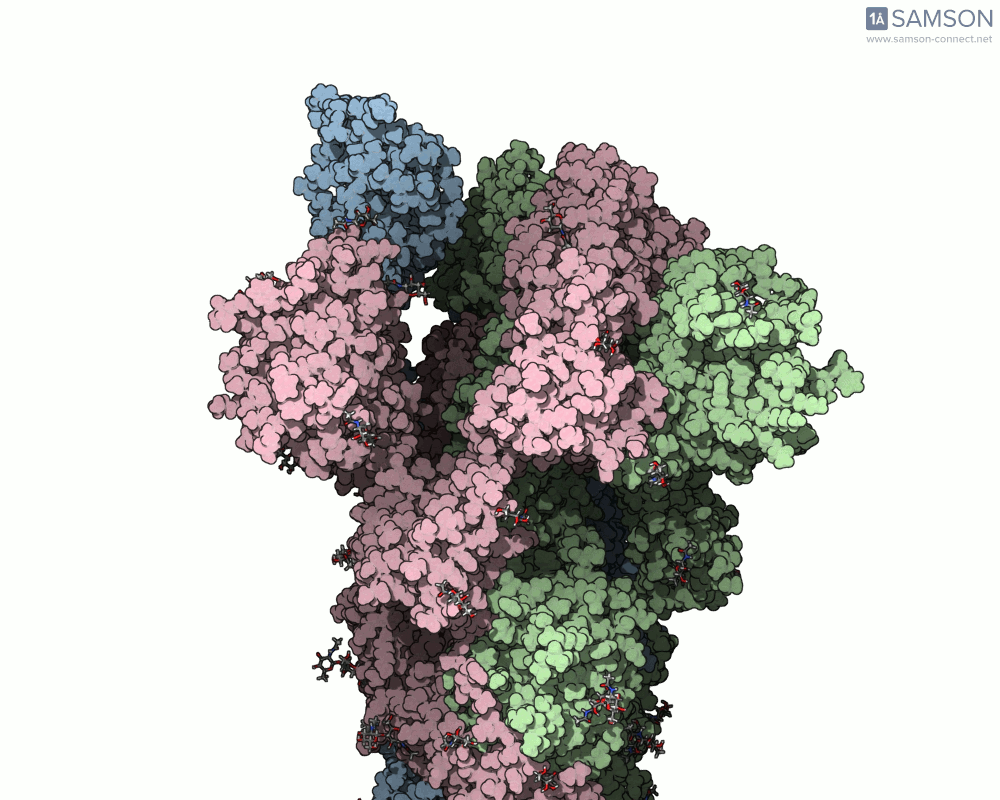
If you’re working in molecular modeling and need to simulate conformational changes, this pipeline offers a practical and efficient method. By leveraging SAMSON’s ARAP and P-NEB modules, you can streamline motion computation for similar biomolecular systems.
Learn more
If you’d like to dive deeper into how this workflow was implemented, including access to code, modules, and other resources, read the full documentation page here: original documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.