





Exploring the effects of small chemical modifications on a lead compound is a daily task for many molecular modelers, particularly when optimizing a series of analogs for desirable properties like potency, selectivity, or ADMET characteristics. However, this form of structure-activity relationship exploration can quickly become tedious without the proper tools. Fortunately, SAMSON’s SMILES Manager introduces a streamlined approach to positional analogue scanning, allowing users to effortlessly generate and analyze analogs by replacing or attaching functional groups at defined positions.
In this post, we will walk through how to identify positions in a molecule and systematically alter them – a common workflow in early-stage drug design – using SMILES Manager. This approach can help you evaluate how different substituents impact your compound’s behavior before moving forward to more computationally intensive steps like docking or quantum-level modeling.
Step 1: Pattern Selection Using SMARTS
After initializing your molecule in SAMSON, either by entering a SMILES string or selecting the molecule in your workspace, the next step is identifying the substructure pattern you want to modify. For example, you might be interested in replacing every aromatic hydrogen in a ring system. To do this, enter the SMARTS pattern [cH] in the designated SMARTS search field.
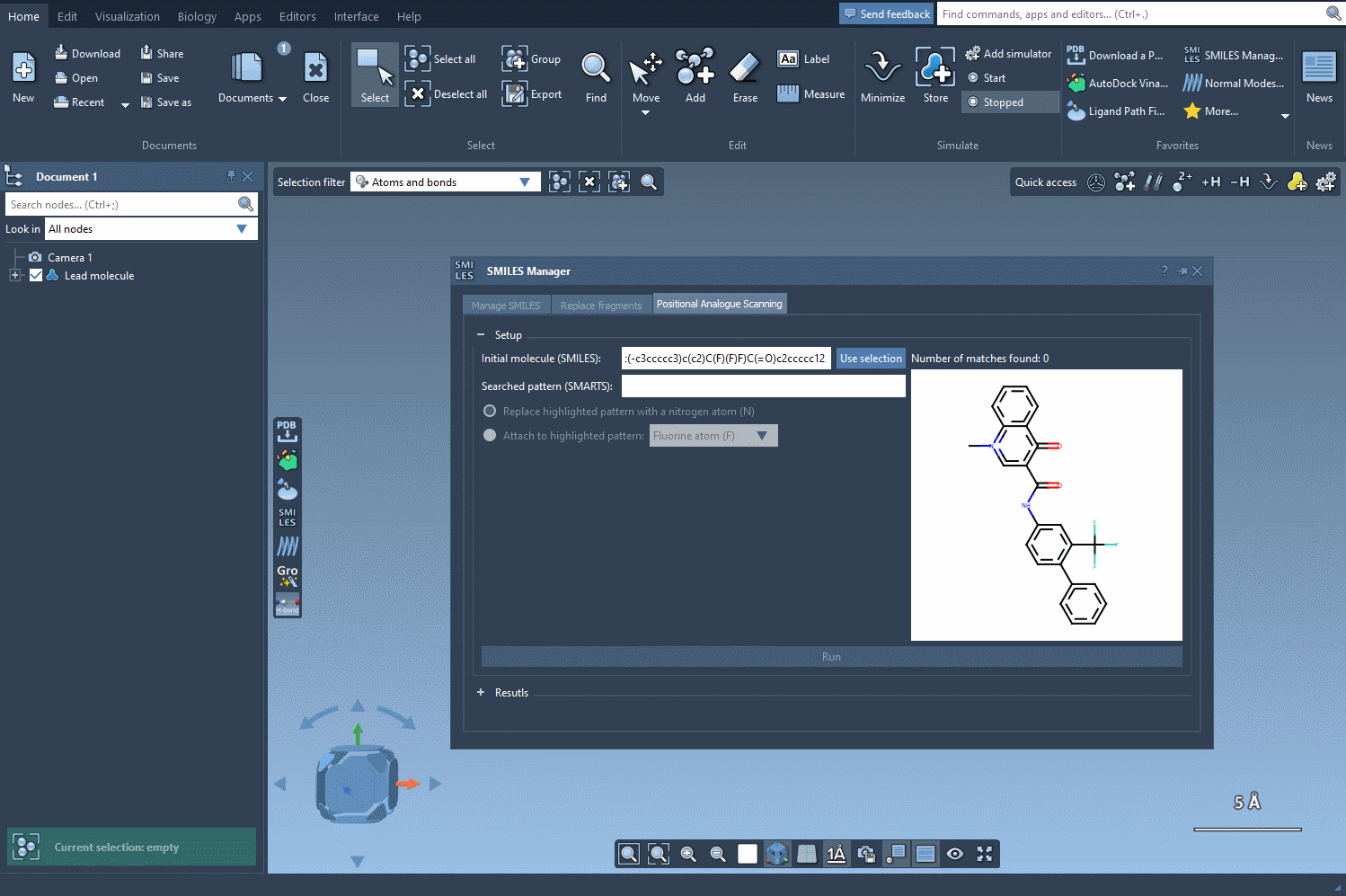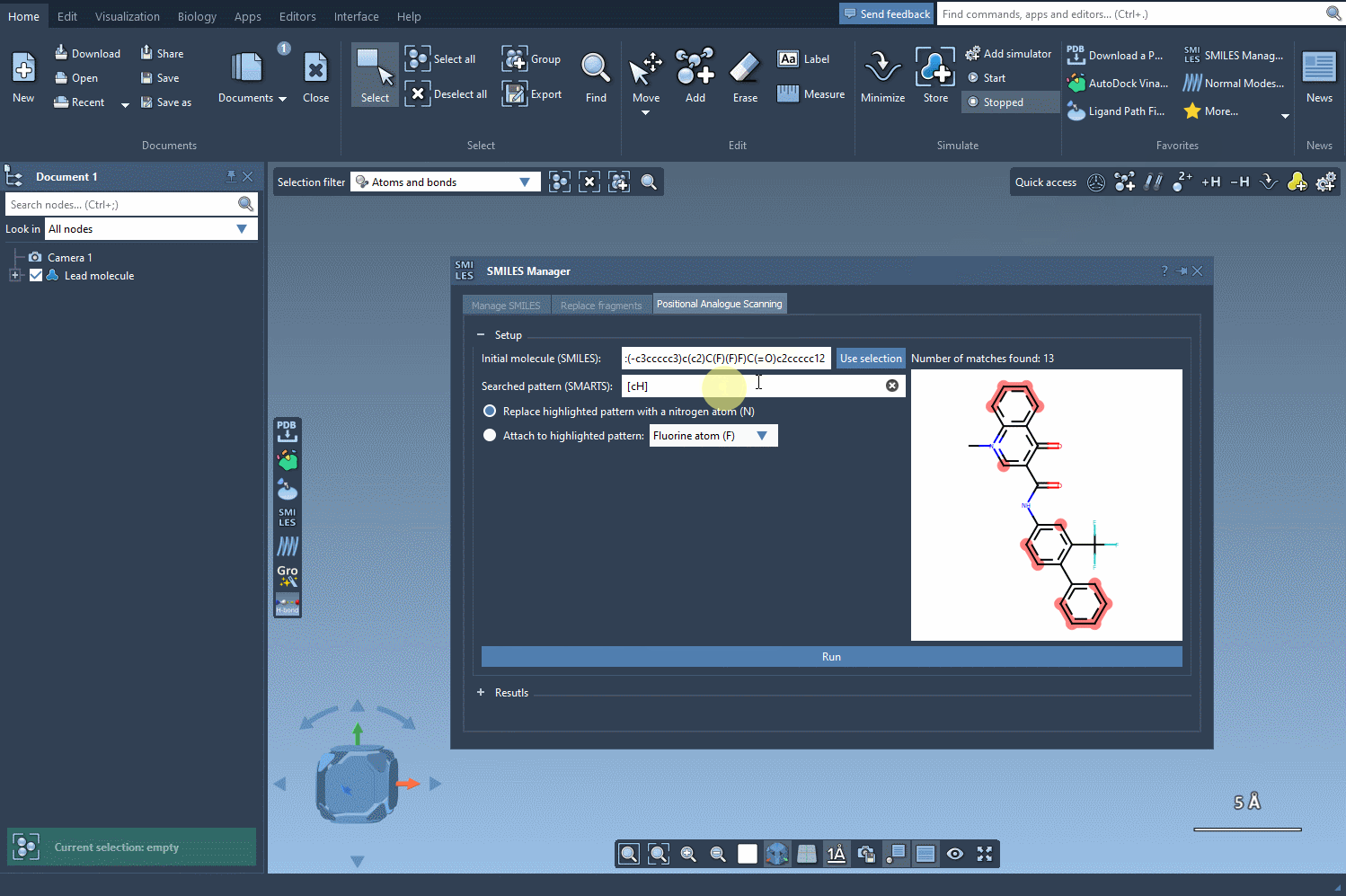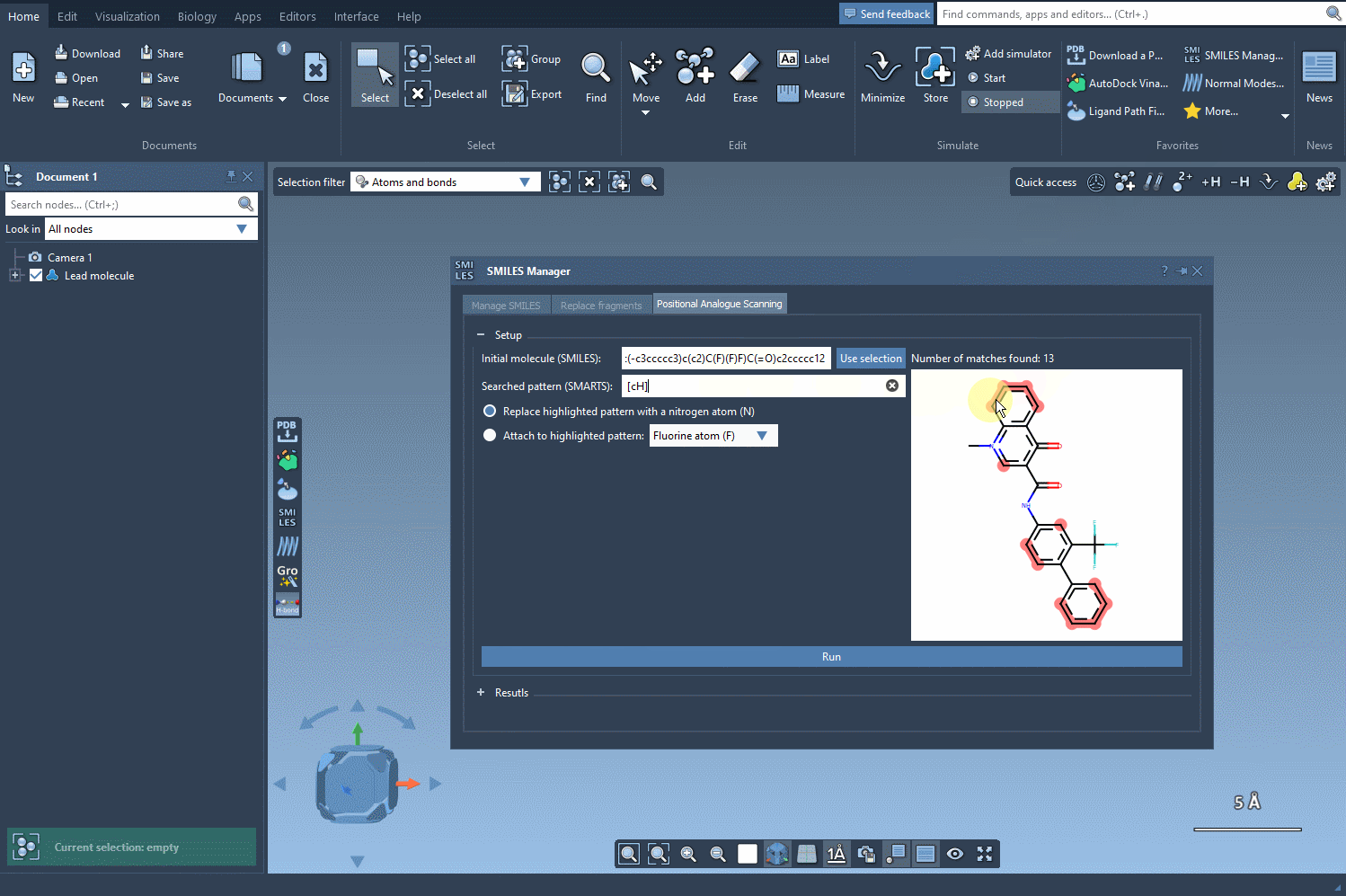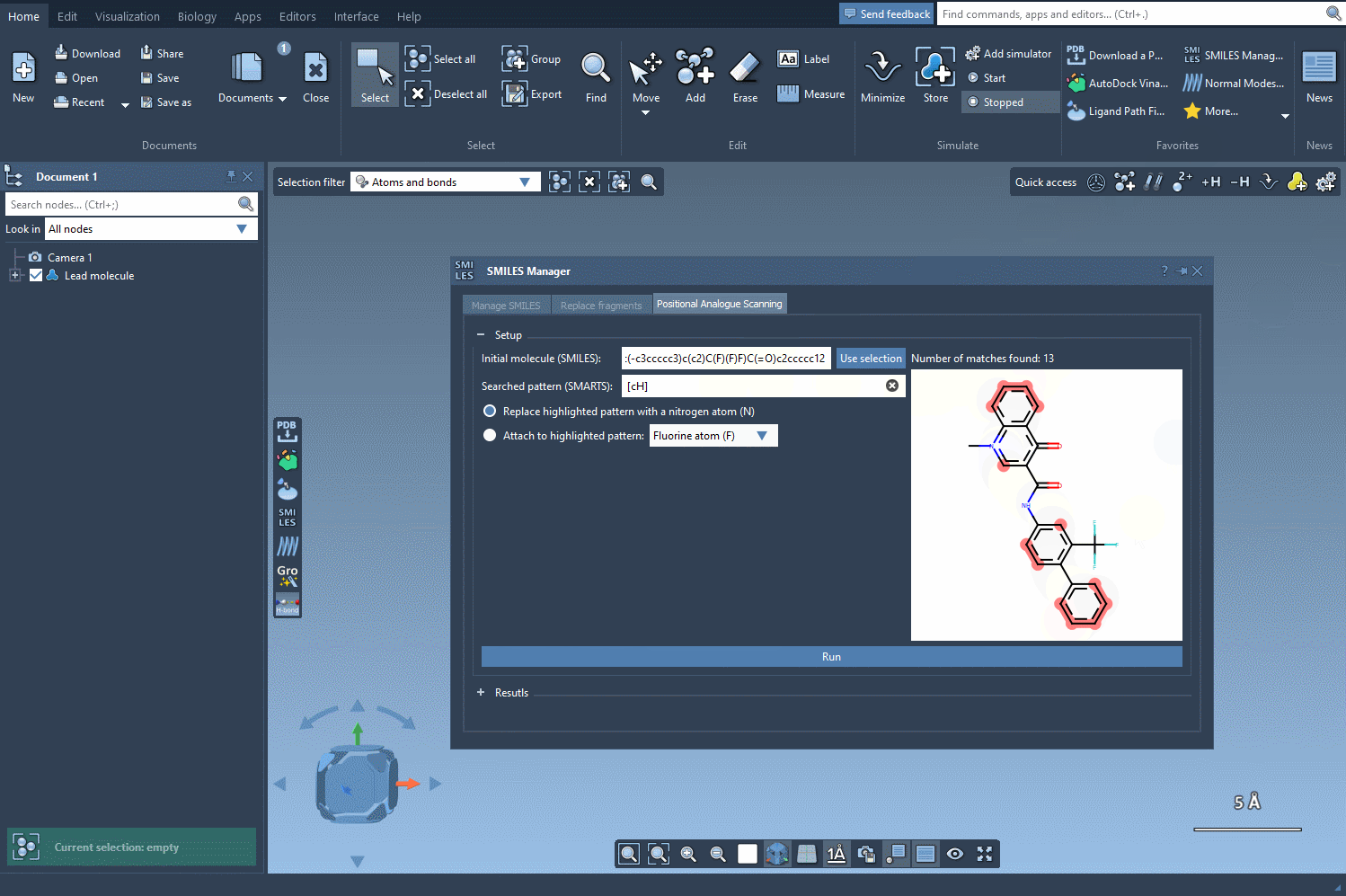
SAMSON will highlight each occurrence of that pattern in your structure, showing where modifications can be applied. This capability gives you precise control over where to perform substitutions—an essential feature when you want to modify specific regions of the molecule while preserving key pharmacophores or functional groups.
Step 2: Replace or Attach New Groups
Once the substructure pattern has been identified, the next step is choosing what to do with it. You can either:
- Replace the identified atoms with a single atom like nitrogen (
N), or - Attach a group such as fluorine (
F) or methyl (CH3).
With a single click on the Run button, SMILES Manager generates a series of analogs based on your input. These are automatically visualized in a 2D format along with their corresponding SMILES strings.
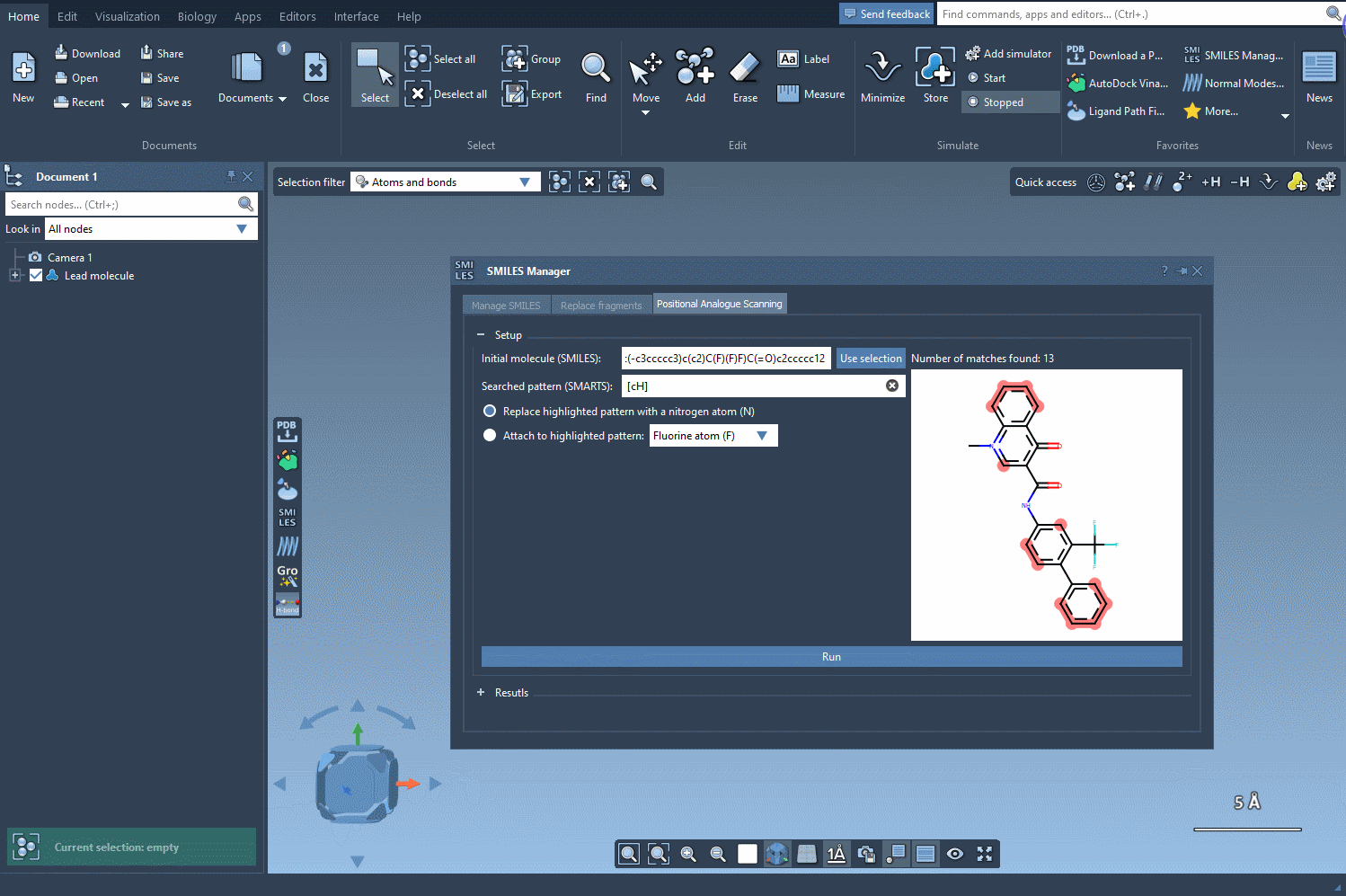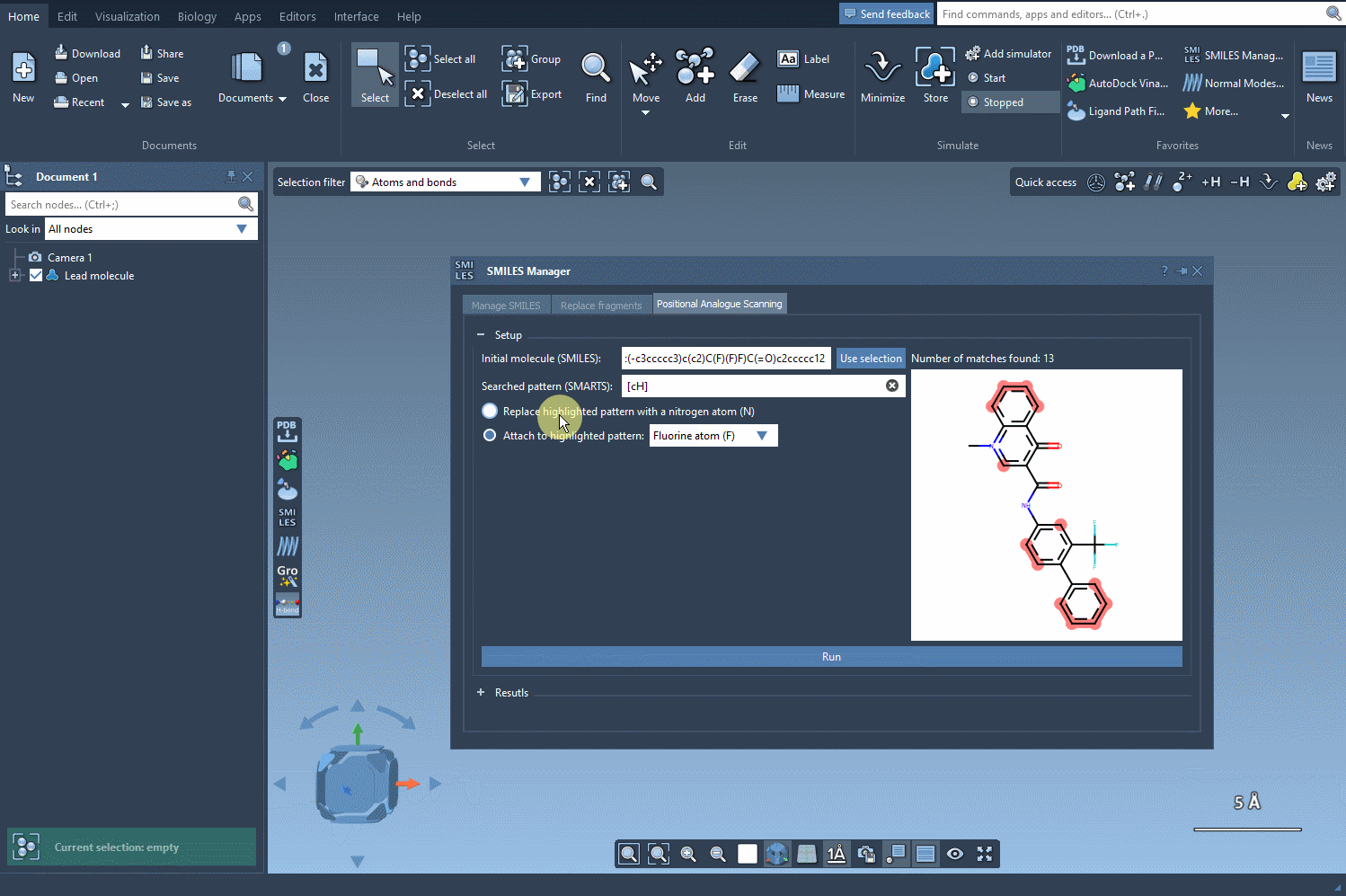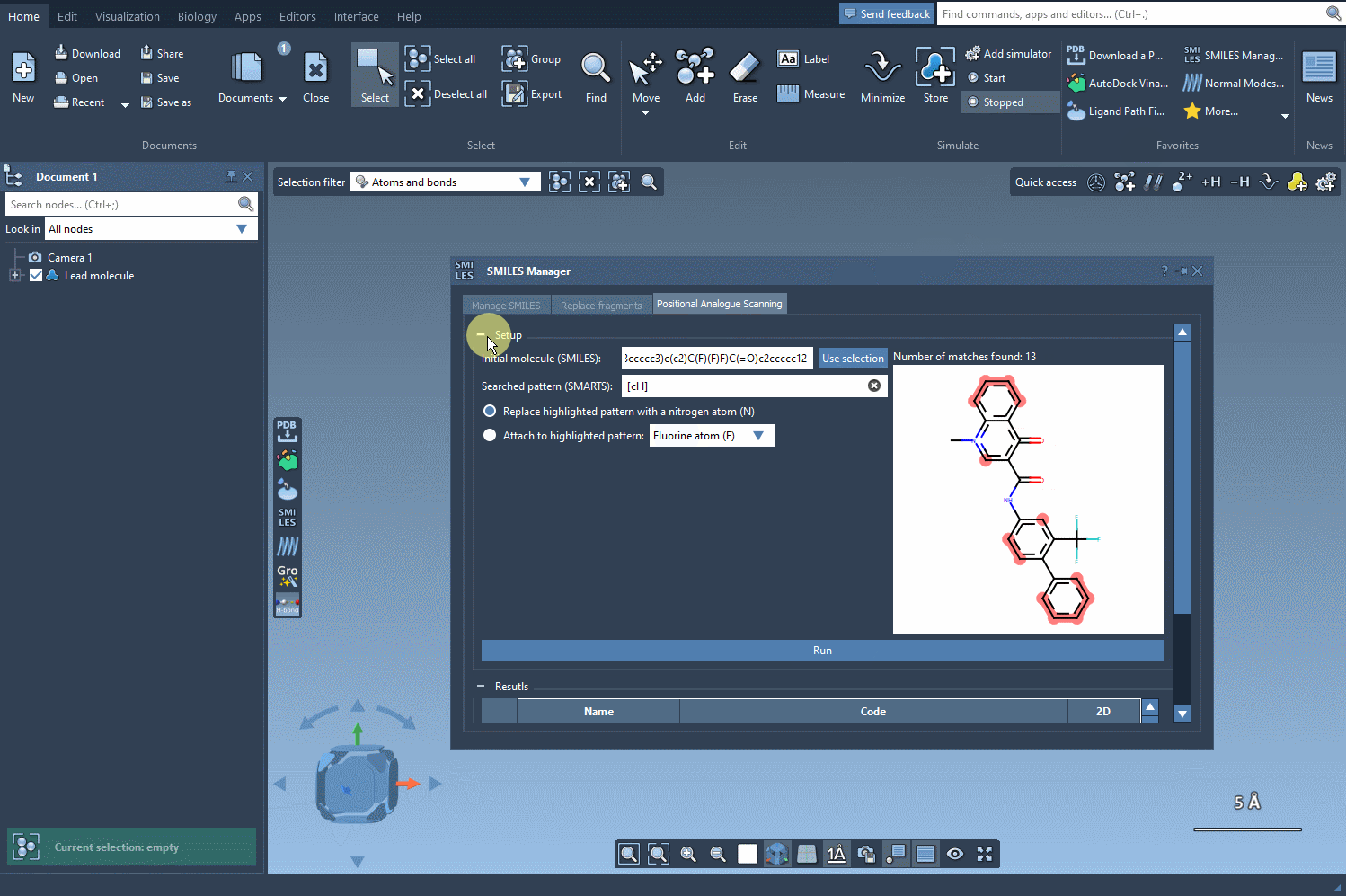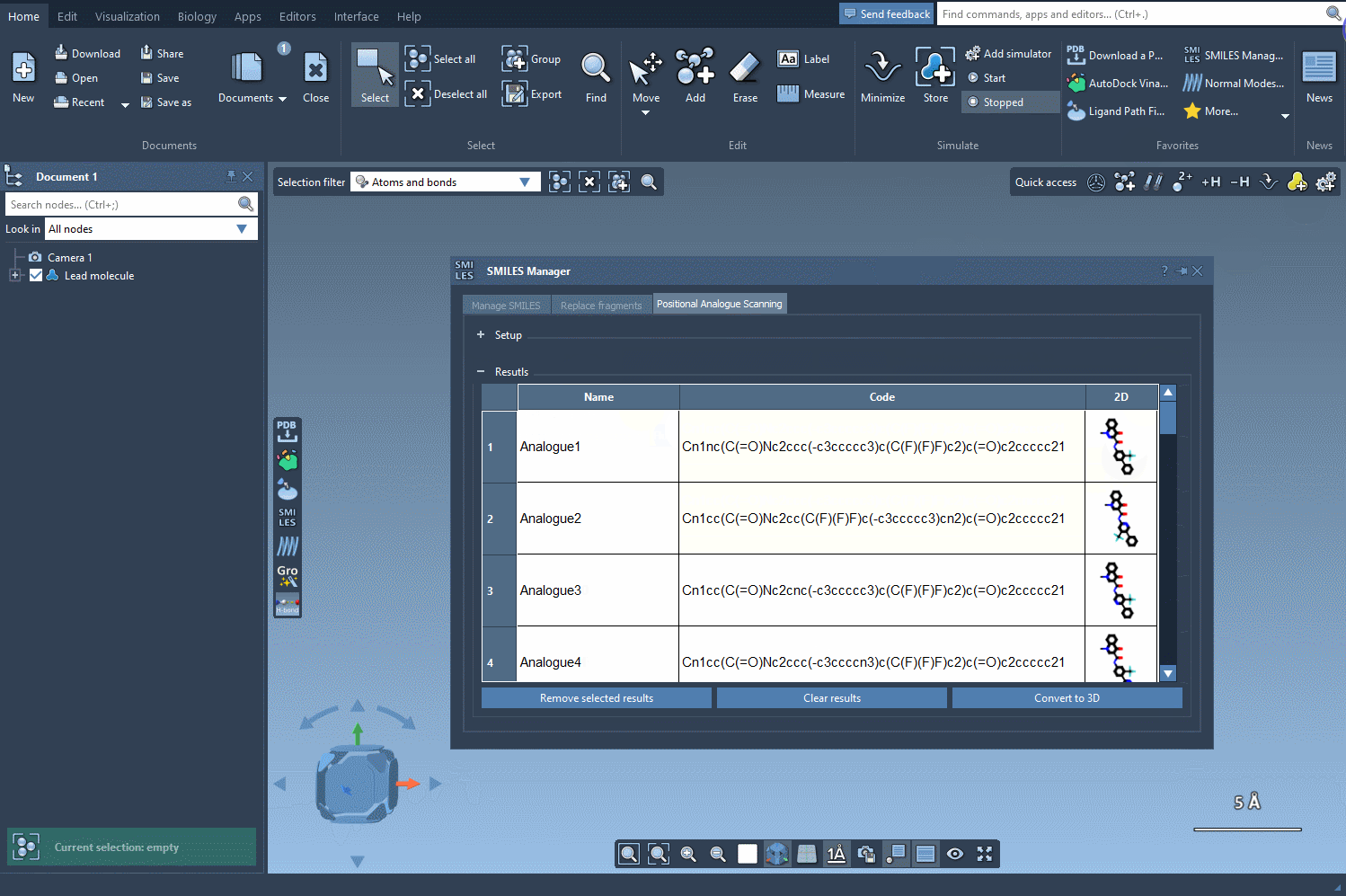
This hands-on flexibility enables you to scan through various substitutions quickly. It’s especially useful when building SAR tables or preparing for docking experiments, as it lets you focus your attention on compounds that show meaningful hypothetical variation.
Why This Matters
When exploring chemical space, the difference between a promising lead and an inactive one can hinge on a single atom. The pairwise comparisons enabled through positional analogue scanning help modelers efficiently explore these subtleties. Instead of manually editing molecules or relying solely on command-line tools, SMILES Manager provides a visual and interactive pipeline that simplifies the process.
Even better, the generated analogs are ready for downstream analyses: you can convert them to 3D structures and dock them directly into your target of interest using the Autodock Vina Extended extension, also within SAMSON.
Final Thoughts
This approach combines chemical intuition with automation, helping you iterate faster while maintaining control over the structural hypotheses you explore. Whether you’re refining a binding pocket interaction or just brainstorming plausible derivatives, positional analogue scanning in SMILES Manager is a helpful, low-friction companion.
To learn more about the full analogue scanning workflow and advanced configurations, visit the official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.