





Designing new molecules often begins with a single starting point: your molecule of interest. But turning that molecule from a theoretical representation into a ready-to-edit structure in a 3D design environment can take time — unless you’re using the SMILES Manager in SAMSON. This post walks you through how to get started quickly by initializing molecular structures from SMILES strings or from your selection in the SAMSON workspace.
Why Initialization Matters 🧲
Before you can analyze, modify, or simulate a molecule, you need to get it into a usable format. While many tools require a series of conversion steps from SMILES to 3D files, SAMSON simplifies the process. Within seconds, you can input your SMILES or select an existing molecule and begin making modifications, running docking simulations, and more — all within the same platform.
Two Ways. One Goal. 🔬
Here are the two main ways modelers can define their starting molecule in the SMILES Manager extension:
- SMILES Input: Paste your SMILES code directly into the interface. This is handy when working from literature or a database.
- Use Selection: If your molecule is already in your SAMSON document, simply select it and click the Use selection button to import it into the SMILES Manager.
This quick-start approach helps eliminate friction, especially when exploring multiple lead compounds or tracking SAR (Structure–Activity Relationship) trends across series of analogs.
Here’s how it looks in action:
Practical Example 🧪
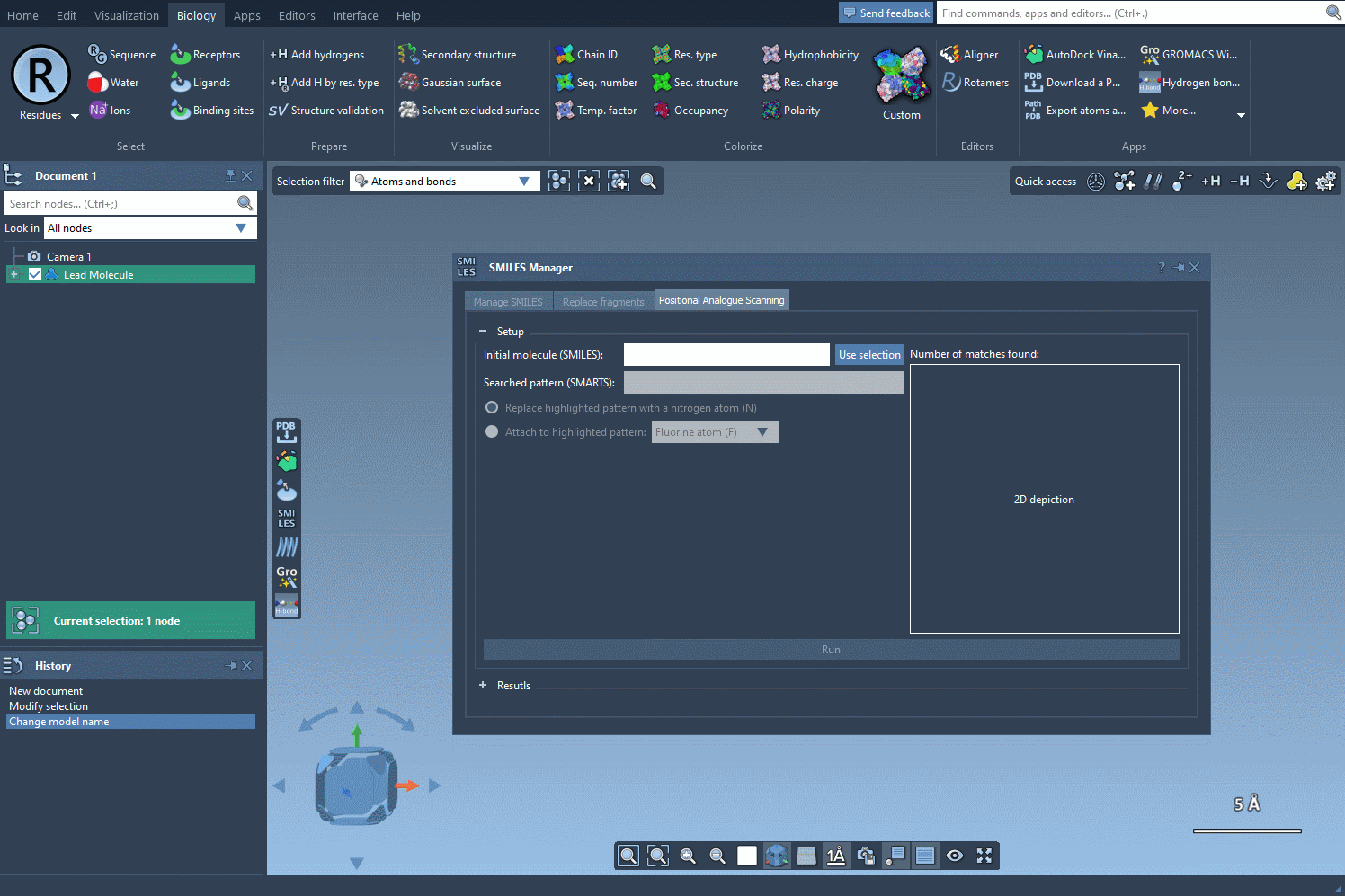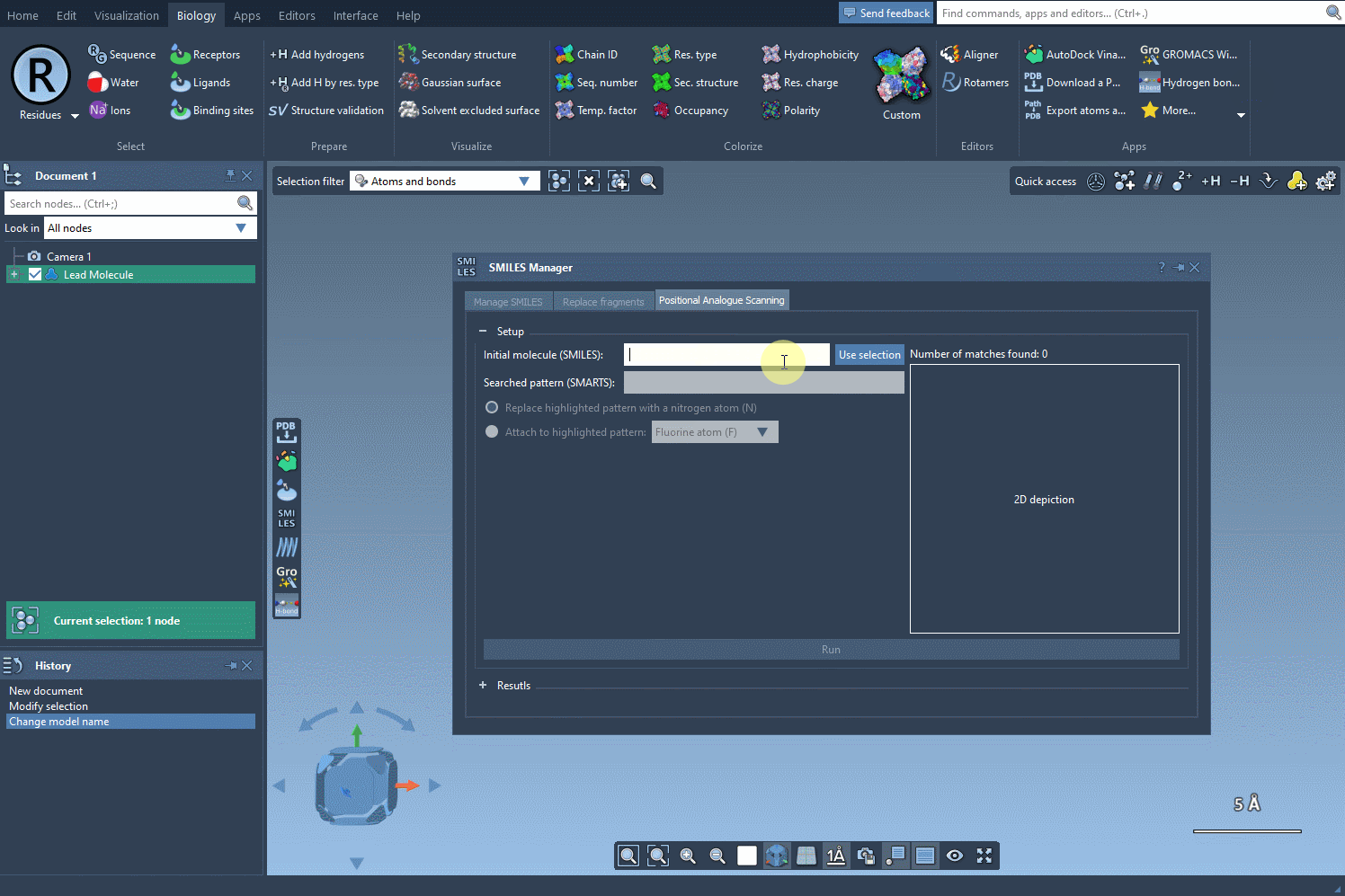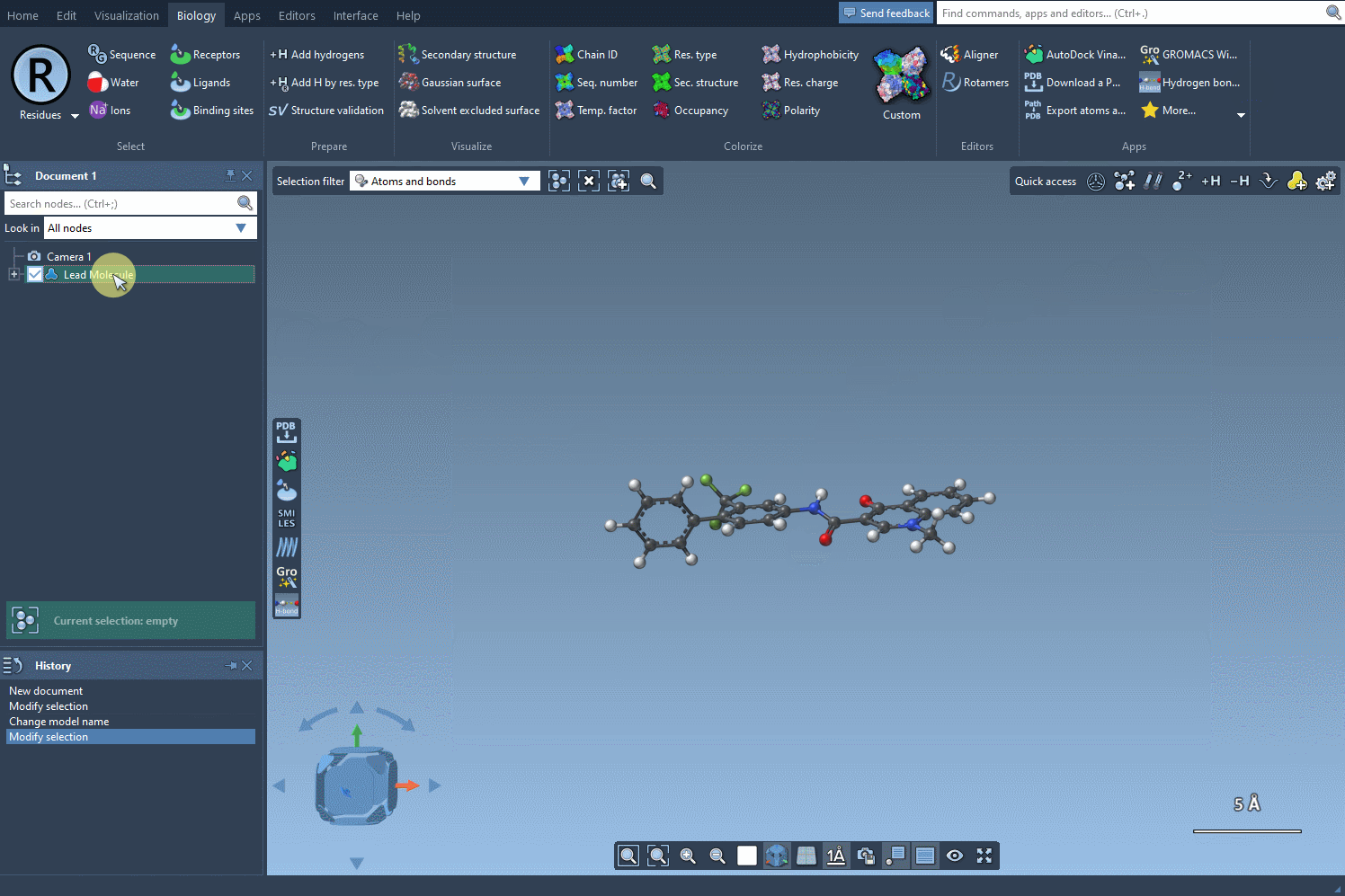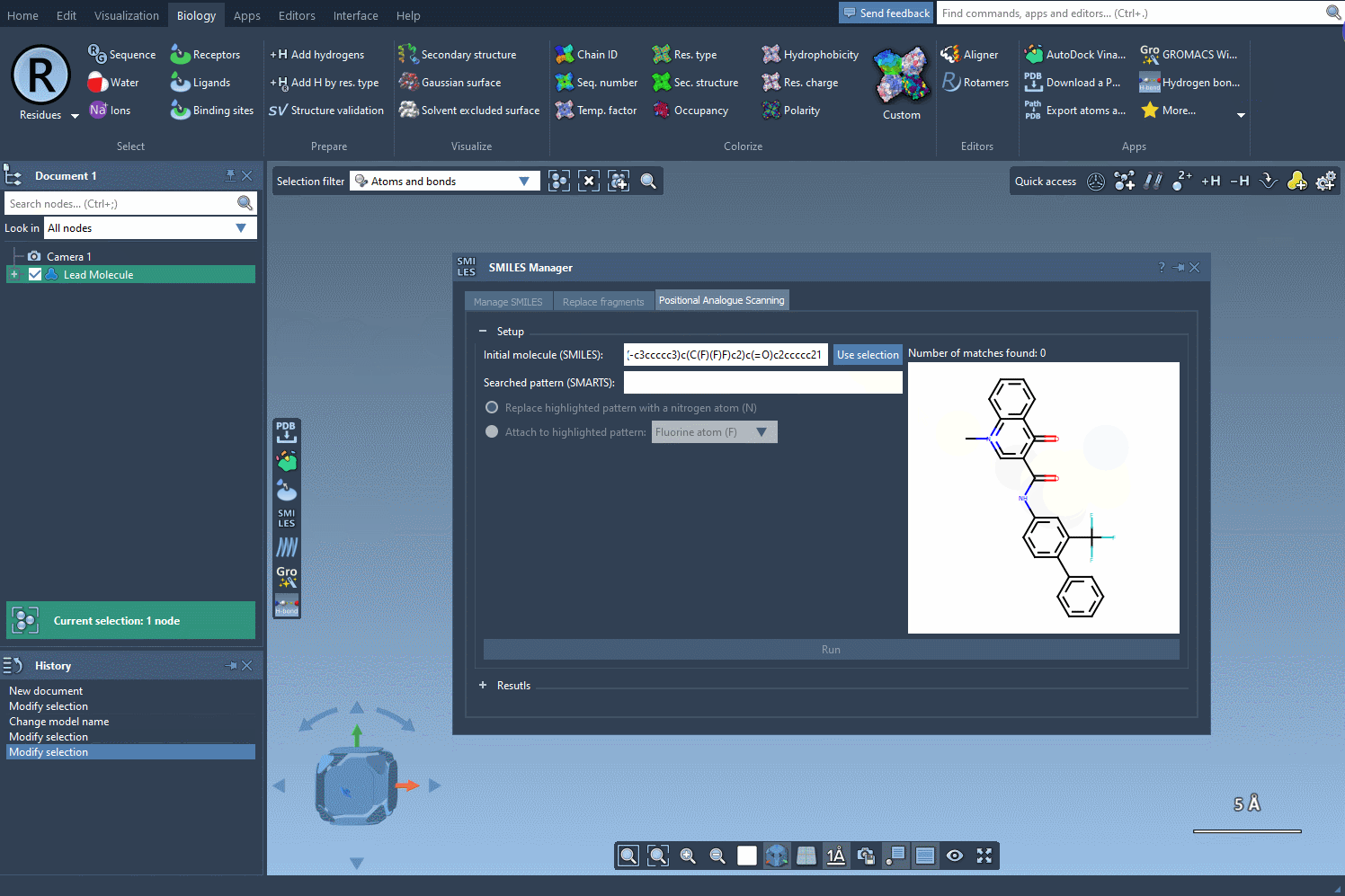
Say you’re working with a molecule commonly cited in literature, such as the following:
|
1 |
CN1C=C(C(=O)Nc2ccc(-c3ccccc3)c(c2)C(F)(F)F)C(=O)c2ccccc12 |
This SMILES string represents a compound studied for its binding properties. Simply paste it into the SMILES Manager, and a 2D structure is instantly generated, visualized, and ready for further actions like analogue scanning or 3D conversion.
Time-Saving for Deeper Work ⏱️
No more jumping between file converters, viewers, or chemical editors. The tight integration inside SAMSON helps keep your focus on design and analysis instead of setup. And because the SMILES Manager supports preset transformations and docking integrations, your journey from initial molecule to drug-like candidate becomes much smoother.
If you’re coming from a medicinal chemistry or structural biology background and want a frictionless way to begin molecular exploration, this feature is worth trying.
To learn more about this and related features, visit the full tutorial at this official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.