





When exploring chemical space for drug discovery or materials design, molecular modelers often face a common issue: how can you systematically explore the effect of small, targeted modifications to a molecule, such as replacing or attaching functional groups at specific sites?
This is where positional analogue scanning comes into play—a practical technique for generating new analogs by modifying defined parts of a molecule. In this blog post, we focus specifically on one of the most powerful yet often overlooked steps in this process: defining the pattern you want to modify using SMARTS codes in the SMILES Manager extension of SAMSON.
Why it matters
When working with substitution patterns—particularly during lead optimization—being able to accurately select the parts of a molecule to scan is crucial. Manual editing can be tedious and error-prone. By using SMARTS patterns (a flexible language for specifying substructures), you can easily define what atoms or groups to target across multiple molecules or analogs.
How to define a pattern using SMARTS
Suppose you’re working with a molecule that contains multiple aromatic rings and you’re interested in exploring what happens when you substitute one of the hydrogens in those rings with a nitrogen atom, a fluorine atom, or a methyl group.
In the SMILES Manager, you can specify the substructure you want to target using a SMARTS pattern. For example, [cH] corresponds to an aromatic carbon with a hydrogen attached (i.e., a common substitution site in aromatic rings).
Here’s what the workflow looks like in practice:
- Enter your molecule of interest via its SMILES code or select it in your document.
- In the SMARTS field, input the pattern you want to modify—e.g.,
[cH]. - The matching atoms in your molecule will be automatically highlighted, as shown below:
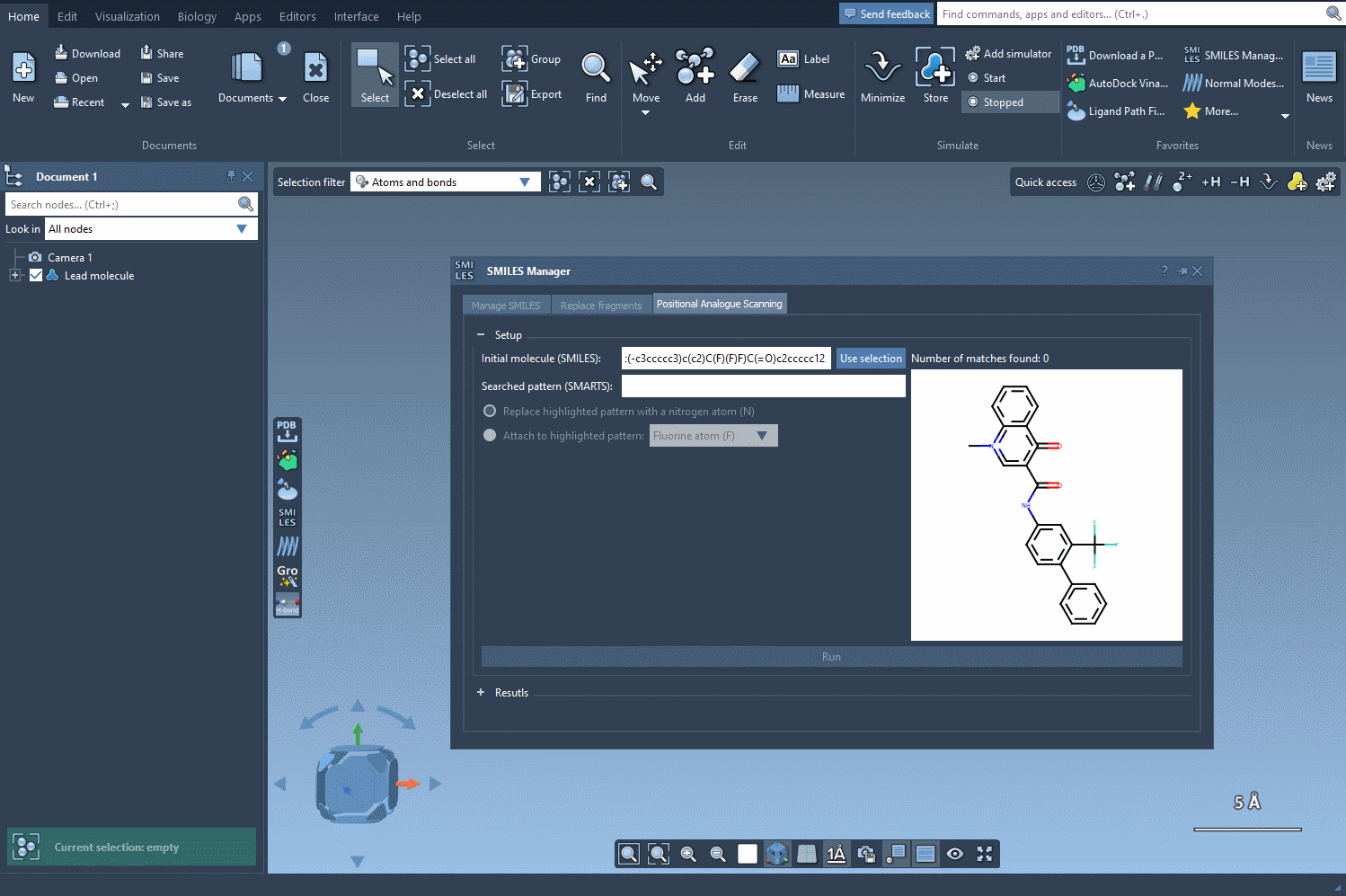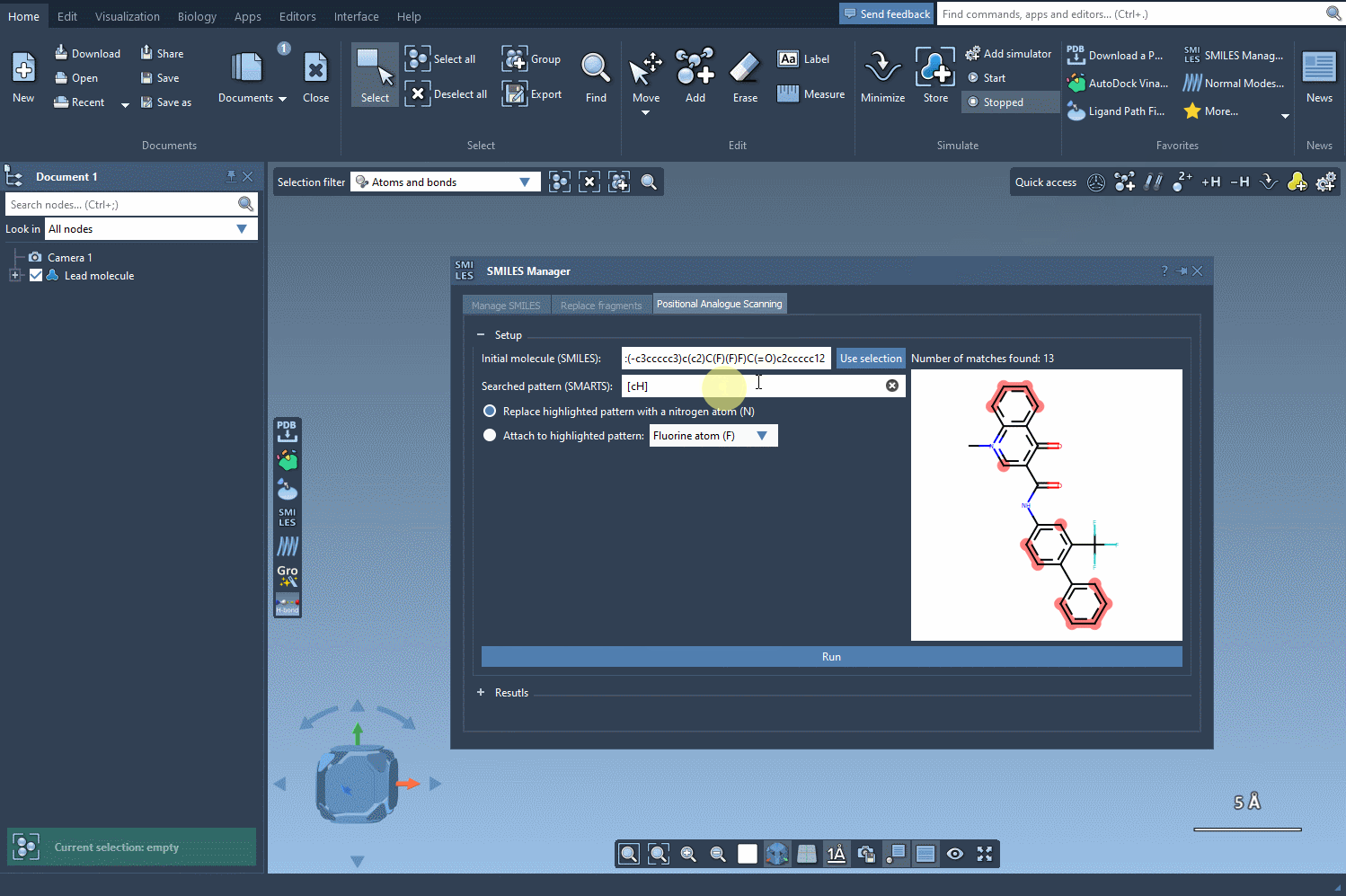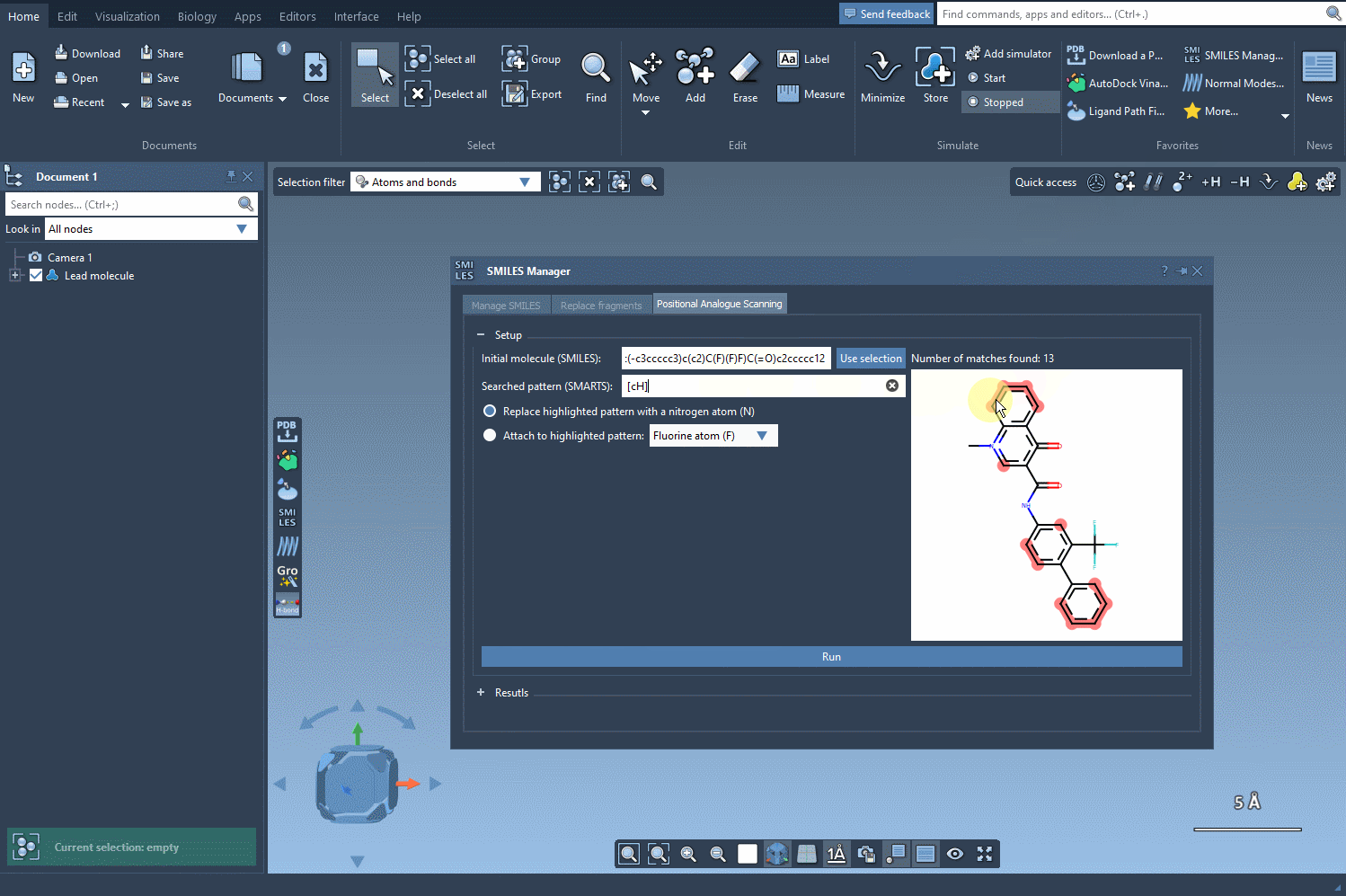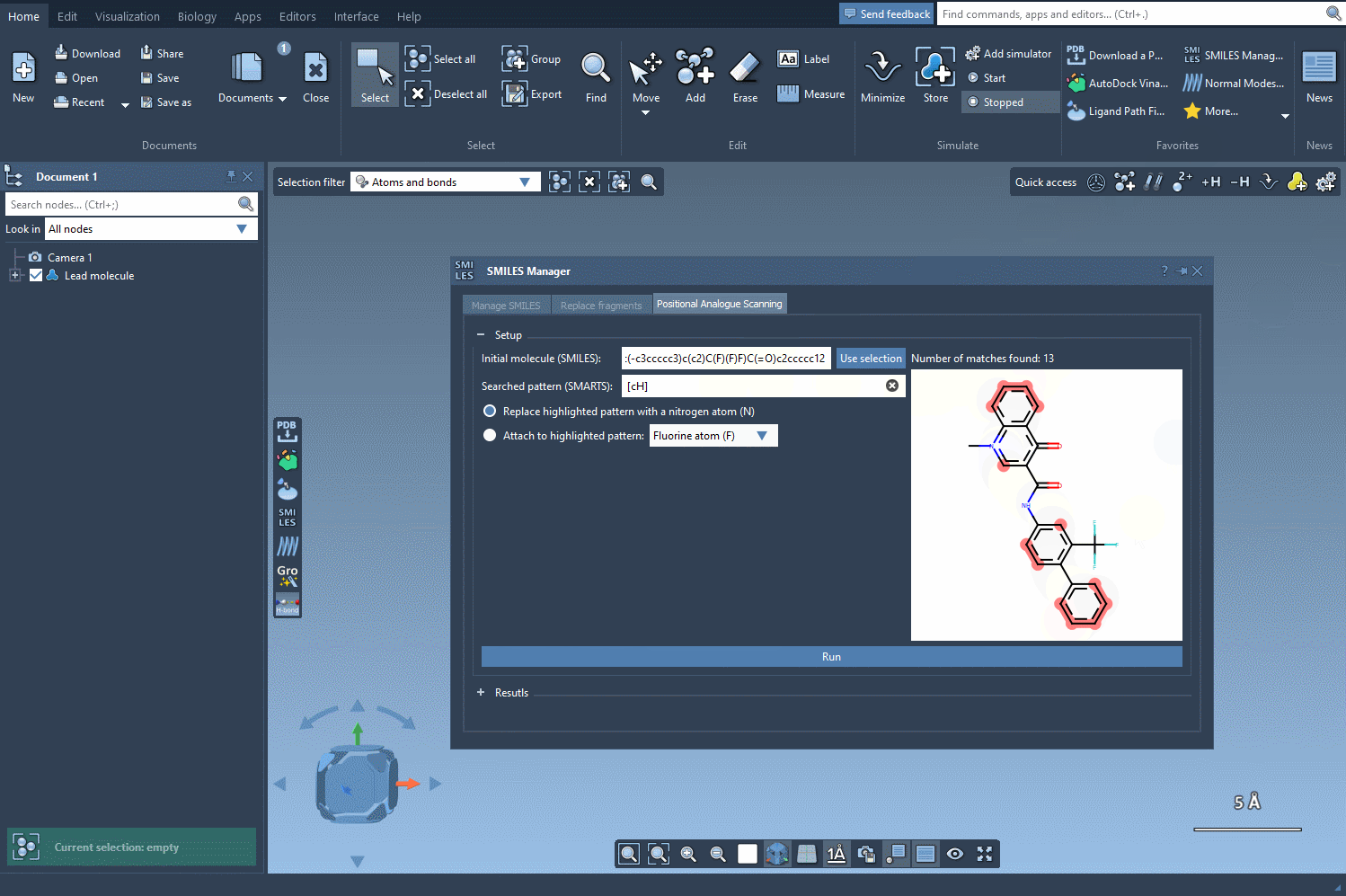
Best practices when choosing SMARTS patterns
Here are a few tips that can help you get the most from your analogue scanning:
- Be specific: Narrow patterns like
[cH]or[nH]provide targeted substitutions and reduce the number of undesired analogs. - Explore different contexts: Try patterns like
c1ccccc1(benzene ring) or[CH3](methyl group) to test larger modifications. - Preview first: After entering a SMARTS pattern, check that the highlights match the part of the molecule you want to explore.
Advantages of this approach
With the use of SMARTS codes, you can design scans focused on chemically meaningful positions, guiding your structure-activity relationship (SAR) analysis more effectively. This minimizes unnecessary computations and focuses your modeling on the changes that matter most.
What’s next?
Once your patterns are defined, you can move on to replacing or attaching different atoms or groups and generating analogs with a single click. These analogs can later be evaluated in 3D, docked into targets, or submitted for property prediction—all within SAMSON.
Ready to start defining your own scanning patterns? You can explore more or follow along with the full tutorial by visiting the official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.