





When you’re exploring structure-activity relationships or looking to modify chemical structures to improve binding affinity, identifying reactive or modifiable sites is often one of the first steps. But this task—searching and highlighting specific substructures—can be repetitive and error-prone, especially when handled manually using traditional cheminformatics tools.
If you’ve ever asked yourself: “Where can I chemically tweak this molecule without disturbing its core structure?”—then SAMSON’s SMILES Manager offers a flexible and efficient way to pinpoint these modification sites using pattern-based selection with SMARTS strings.
Why pattern selection matters
When performing positional analogue scanning, chemists systematically vary atoms or groups at particular positions in a molecule to explore effects on activity. SMARTS (SMiles ARbitrary Target Specification) allows you to express complex substructural patterns and find all matching atoms in a molecule, whether it’s an aromatic carbon, a particular side chain, or even a specific hybridization.
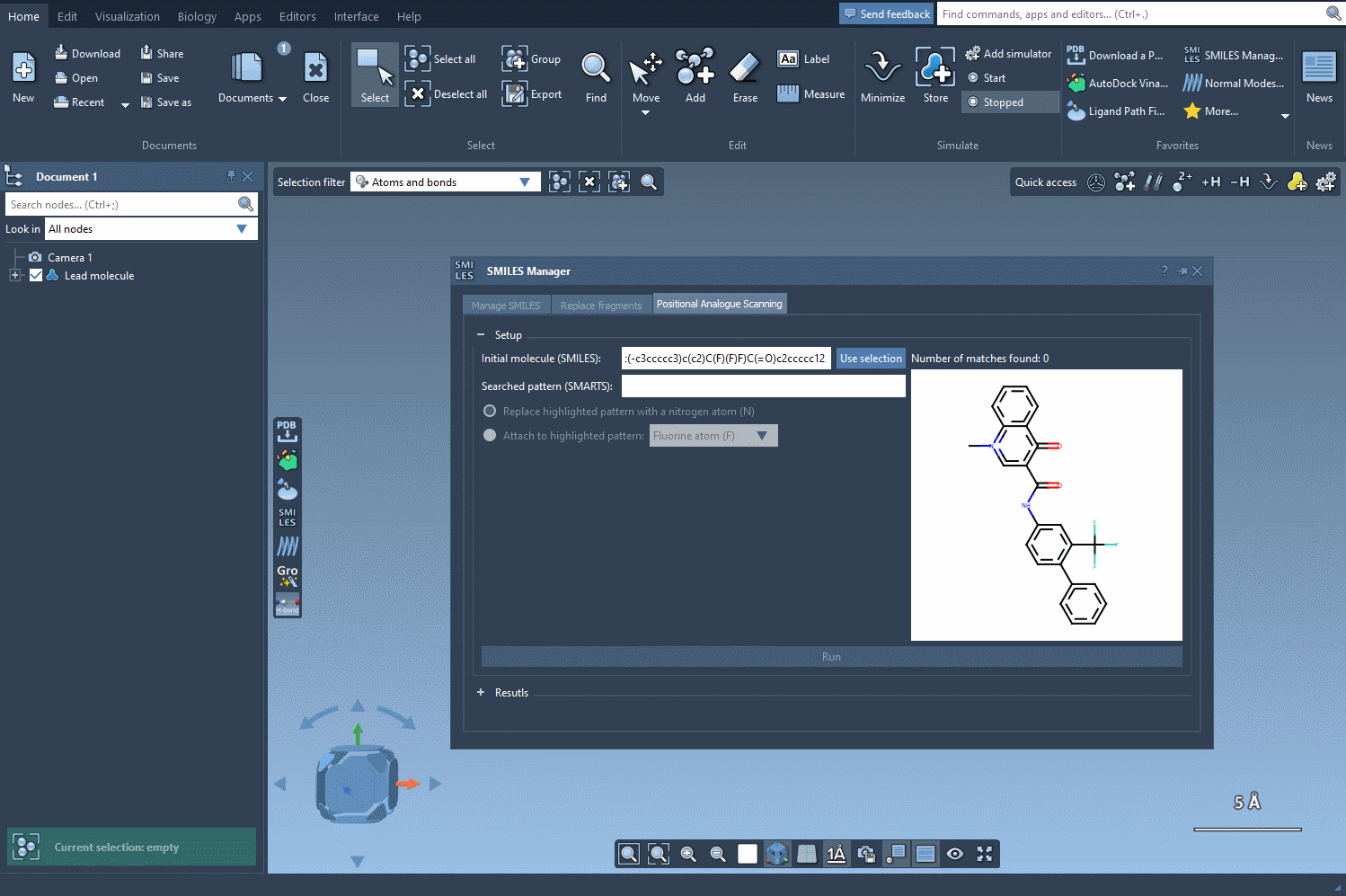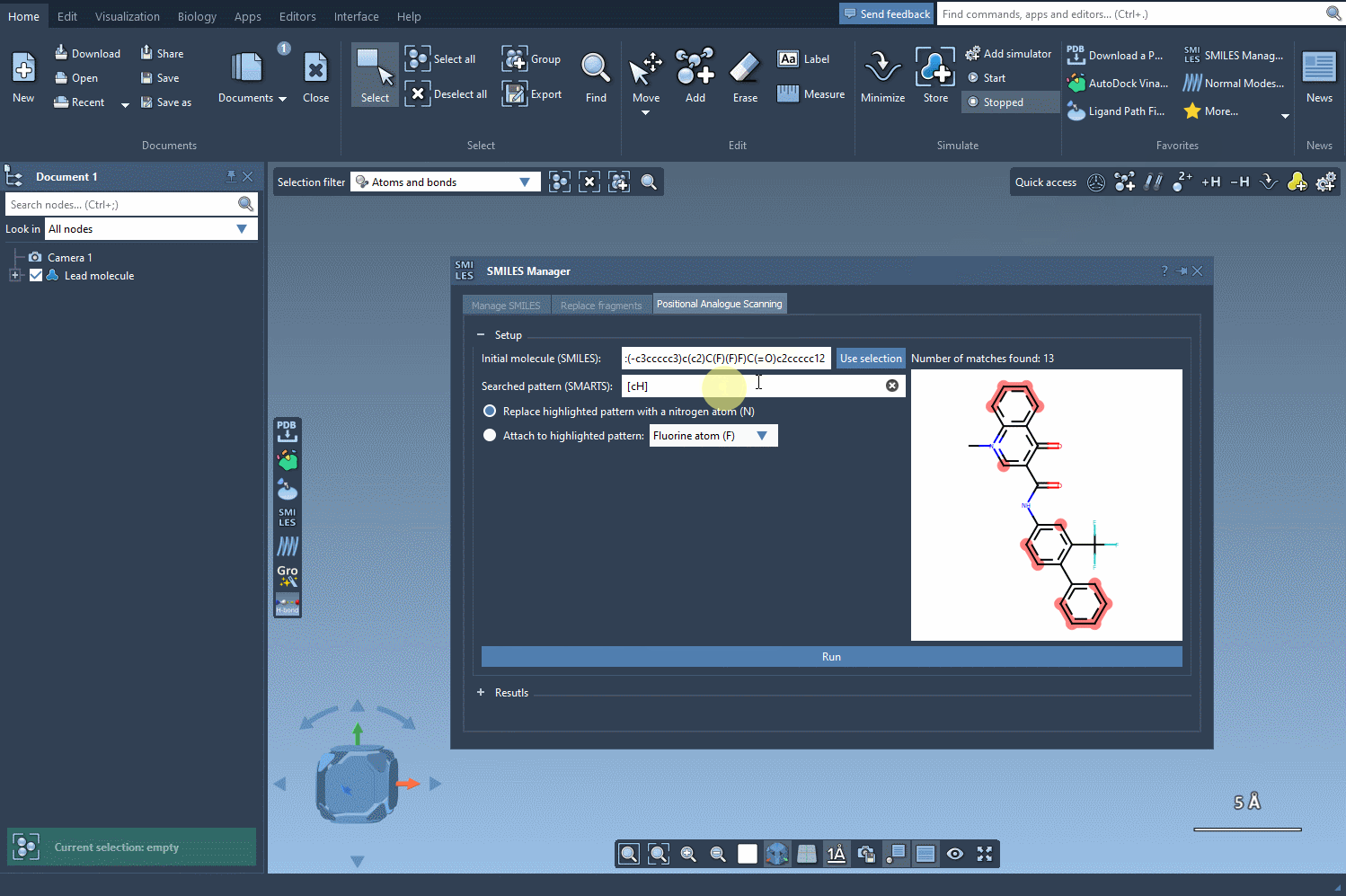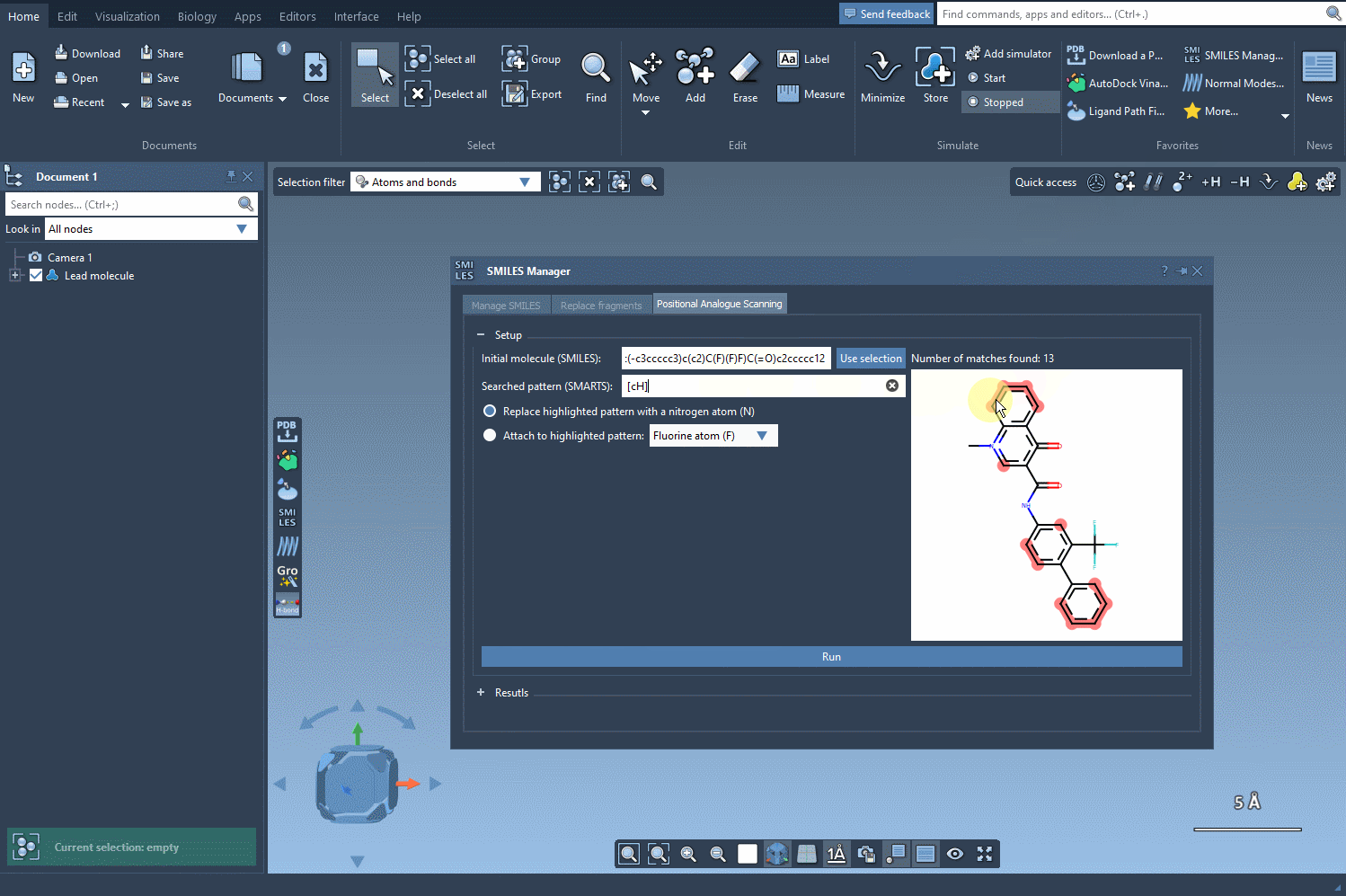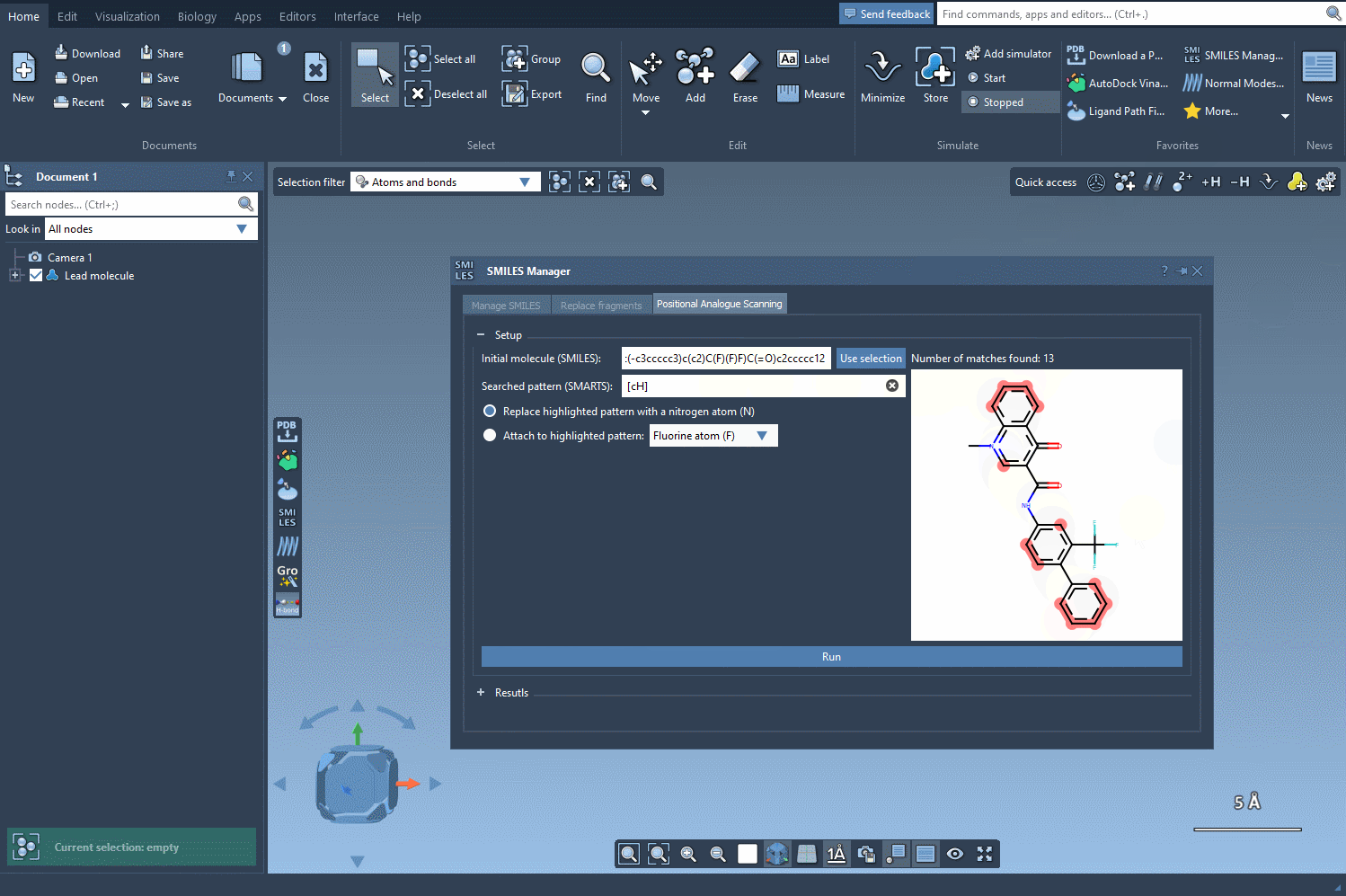
With SMILES Manager in SAMSON, you can begin by entering the SMARTS code [cH] to select protonated aromatic carbons. Once entered, the matching atoms in your molecule are automatically highlighted—instantly showing you all possible points for substitution or addition. This visual feedback reduces ambiguity and helps ensure you don’t miss key positions in the structure.
How it works, step by step
- Start by loading your molecule in SAMSON. You can either paste its SMILES code or select it from the workspace using the Use selection button.
- Switch to the SMARTS input field and enter your desired pattern. For aromatic carbons, use
[cH]. The molecules will immediately display highlighted atoms matching this pattern. - Once patterns are identified, choose how you’d like to modify them: substitute or attach new groups, such as replacing a carbon with nitrogen (N) or connecting a methyl group (CH3).
This approach is particularly helpful when working on hit-to-lead optimization where systematic modifications are necessary to probe structure-function relationships. Instead of relying on static diagrams or notes, you interact with the actual molecular environment—gaining intuition and saving time.
Tips for better results
- Be precise with SMARTS patterns. A generic pattern like
[c]may return too many matches. Narrow it down with descriptors like[cH]or[cX3]to focus on likely reaction sites. - Use the animated visual feedback to ensure that your pattern matches what you intend. If you’re seeing too many or too few highlights, refine your SMARTS code.
Fewer clicks, more insight
This visualization of internal chemical logic—from typing a single SMARTS pattern to seeing all modification sites—can be a game-changer in early-stage ligand design. Instead of guessing or relying on static 2D editors, you get immediate feedback and the possibility for rapid iteration within the same molecular environment.
To explore the full workflow, including how to then generate analogs and simulate their behavior via docking, visit the full documentation:
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.