





Superimposing protein structures to compare their folds or map conserved regions is a common task in computational biology. But doing it manually—whether with PyMOL scripts or rigid RMSD calculations—can be repetitive and error-prone. If you’re spending time writing custom scripts each time you want to compare two structures, you might appreciate a visual, interactive alternative.
In this blog post, we’ll walk through how to quickly superimpose two related hemoglobins using the Protein Aligner in SAMSON. This doesn’t require coding and gives you real-time alignment feedback with built-in highlight tools. Whether you’re preparing homology models or investigating structurally conserved motifs, this can save you a lot of time.
Start by Fetching the Structures
Let’s say you want to compare two hemoglobin structures from different organisms, referenced by their PDB codes 1DLW and 1RTX. Here’s how you can load them into SAMSON:
- Open Home > Fetch.
- Enter
1DLW 1RTXin the PDB search bar. - Click Load.

Optional: Clean Up the Structures
Before aligning, consider cleaning the systems to remove water, ligands, or alternative atom locations. This step is helpful to ensure only the relevant parts are aligned.
This can be done via Home > Prepare.
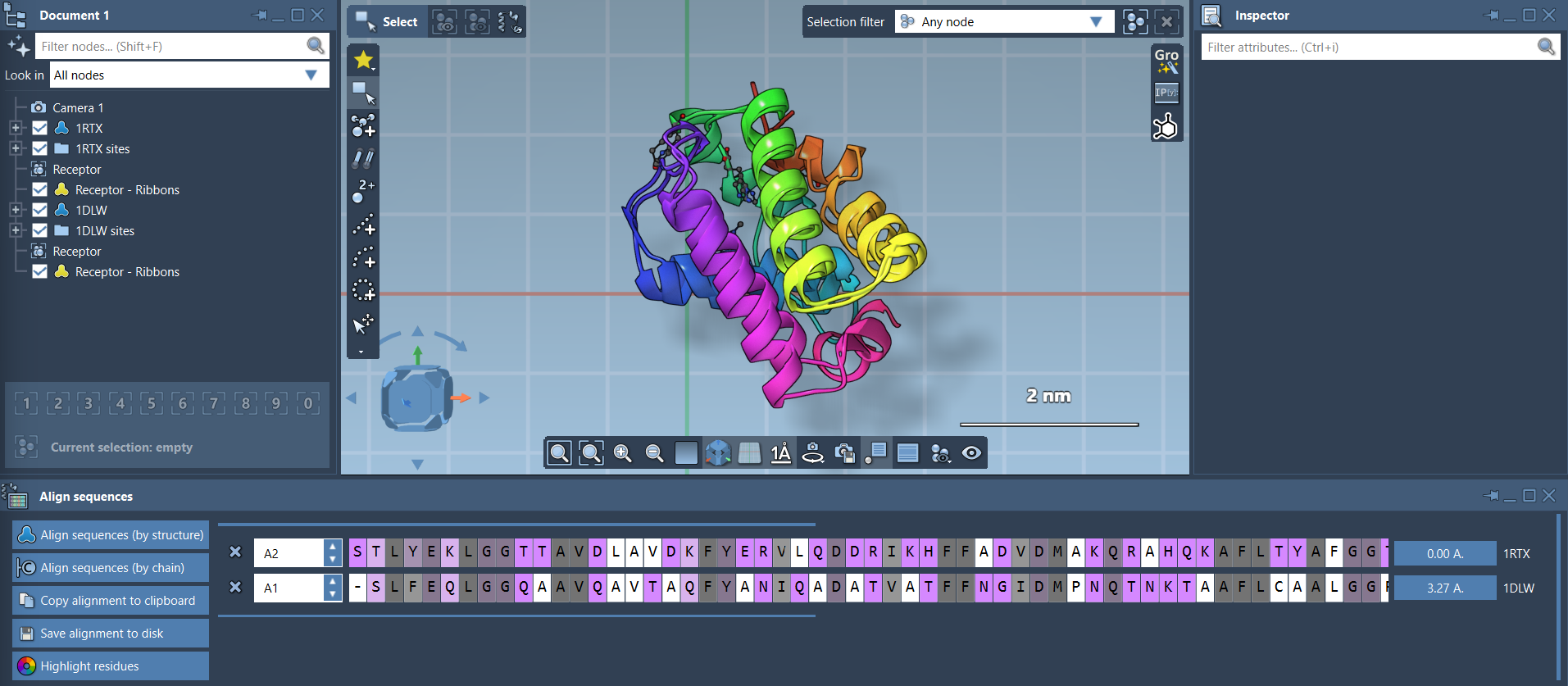
Superimpose the Structures
To launch the Protein Aligner, go to Home > Align and select the extension (a tiny red corner protein icon 🧬).

Once both models are loaded and visible:
- Ensure no residues are selected (click in the viewport to deselect).
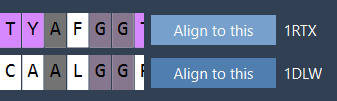
- In the Protein Aligner window, click Align to this on the first model’s row (e.g., 1DLW).
This superimposes the second model (1RTX) on top of the first. The tool automatically computes the RMSD between the aligned structures.

Visualize with Ribbons
If the final alignment looks like a tangle of dots, don’t worry: you can toggle ribbon representations under Visualization > Visual model > Ribbons. To color the two proteins differently, select one first, then apply the visual model. This helps assess alignment effectiveness more clearly.
Focus on Structural Segments
Need to compare a specific region—say, an alpha helix at the N-terminus—but not the whole structure? The Protein Aligner lets you align based on selected residues only:
- Select the first 20 residues in both sequences using the sequence viewer.
- Click the alignment button beside your selection (it may show an RMSD value like
0.0 Å).


Who Is This Useful For?
If you’re working on homology modeling, active site comparison, or tracking structural changes after mutations, this interactive method lets you focus on your actual goal—understanding the biology—rather than waste time adjusting scripts or inputs.
Learn more about the entire alignment process and related tools in the official documentation: Protein Aligner Documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.