





One of the surprisingly common pains molecular modelers face is interpolating the motion between two conformational states of a macromolecular complex when those structures differ in the number of atoms or residues. Even small differences can impede trajectory generation, especially with overlaid visualizations or energetic analysis in downstream applications. This is particularly true when working with flexible complexes like the SARS-CoV-2 spike protein.
So what do you do when your two PDB structures are fundamentally not the same? Do you throw them into a morphing tool and accept strange artifacts? Or do you spend time aligning and pruning the models manually? A better way might be hidden in plain sight.
Case Study: The Opening Motion of the SARS-CoV-2 Spike Protein
Researchers behind the SAMSON molecular design platform have tackled this problem when modeling the transition from the closed state (PDB 6VXX) to the open state (PDB 6VYB) of the SARS-CoV-2 spike protein. The challenge? These two experimental structures differ in the number of residues. Yet the team was still able to interpolate a reliable conformational pathway between them using a clever combination of techniques within SAMSON.
Step-by-Step Workflow
1. Preprocess Structures
Differences in sugar representation and hydrogen atoms could mislead interpolation. So a Python script was used to adjust bond orders in sugar molecules, followed by hydrogenation and energy minimization for both models.
2. Initial Interpolation with ARAP
Using the As-Rigid-As-Possible (ARAP) module in SAMSON, a preliminary path was calculated from the spike’s open state to the closed state. This initial interpolation produced a structurally consistent set of frames — but only for the atom set of the open state (6VYB).
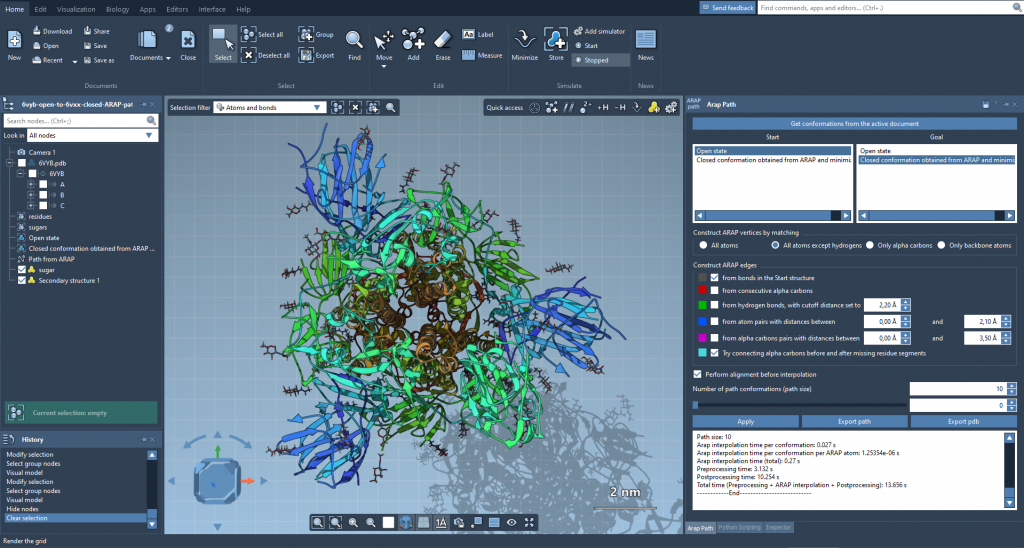
3. Generate Compatible Conformations
Because the ARAP path used 6VYB’s atom set, a key trick was to extract a conformation close to the closed state from this interpolated trajectory. This adjusted version of the closed conformation now had the same number of residues as 6VYB, solving the inconsistency issue.
4. Rebuilding and Further Interpolation
With both start and end conformations now compatible, the ARAP interpolation was re-run using the adjusted closed structure, followed by refinement using the Parallel Nudged Elastic Band (P-NEB) module for more chemically realistic motion.

Why This Matters
This workflow offers a practical solution for molecular modelers who wish to interpolate conformational motion between structurally inconsistent states. Instead of discarding valuable experimental data, you can align your atom sets by generating intermediary states that help reconcile atom counts and connectivity. This is especially valuable for protein-ligand systems, dynamic complexes, or large viral structures where crystal structures vary from source to source.
Bonus: Ready-to-Use Trajectories
For visualization or study, you can also download the resulting interpolated trajectories of the SARS-CoV-2 spike motion from the SAMSON documentation in formats such as PDB (single file) or SAMSON’s native format (.sam), including pre-defined paths and animated loops.
Learn more about this method and the computed spike trajectories in the original documentation: documentation.samson-connect.net
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON from www.samson-connect.net.