





For molecular modelers, one of the recurring challenges involves handling significant topology changes during molecular simulations. Whether it’s forming new bonds, breaking existing ones, or smoothly transitioning between different molecular structures, having a reliable tool to manage these transformations dynamically can make all the difference. The Interactive Modeling Universal Force Field (IM-UFF) extension in SAMSON provides a practical and flexible solution to this problem, allowing users to edit and simulate molecular systems interactively and in real time.
What Makes IM-UFF Special?
Unlike the traditional Universal Force Field (UFF), the IM-UFF interaction model enables smooth topology changes while considering the full periodic table, as originally proposed by Rappe et al. IM-UFF supports:
- Creation and breaking of covalent bonds.
- Changing covalent bond orders dynamically.
- Updating atom typizations automatically as structures evolve.
This capability means that molecular structures can undergo significant modifications during simulation while being guided by physically-based inter-atomic forces. This makes IM-UFF an excellent tool for tasks like simulating chemical reactions, exploring dynamic conformational spaces, or designing new molecular systems interactively.
Setting Up and Running IM-UFF
Getting started with IM-UFF is straightforward. Here’s a quick guide:
- Open a document containing the molecular system you want to simulate.
- Add a simulator via
Edit > Simulate > Add simulator, or use the shortcut: - Windows/Linux: Ctrl+Shift+M
- MacOS: Cmd+Shift+M
- Select Interactive Modeling Universal Force Field from the list of interaction models.
- Choose the state updater (e.g., FIRE) and proceed by pressing OK.
Once configured, you can modify the interactive modeling options in the parameter settings. IM-UFF offers options such as:
- Static topology (UFF only) – Switch between standard UFF and IM-UFF setups.
- Keep vdW for manipulated – Simplify manipulations by excluding the manipulated atoms from van der Waals interactions.
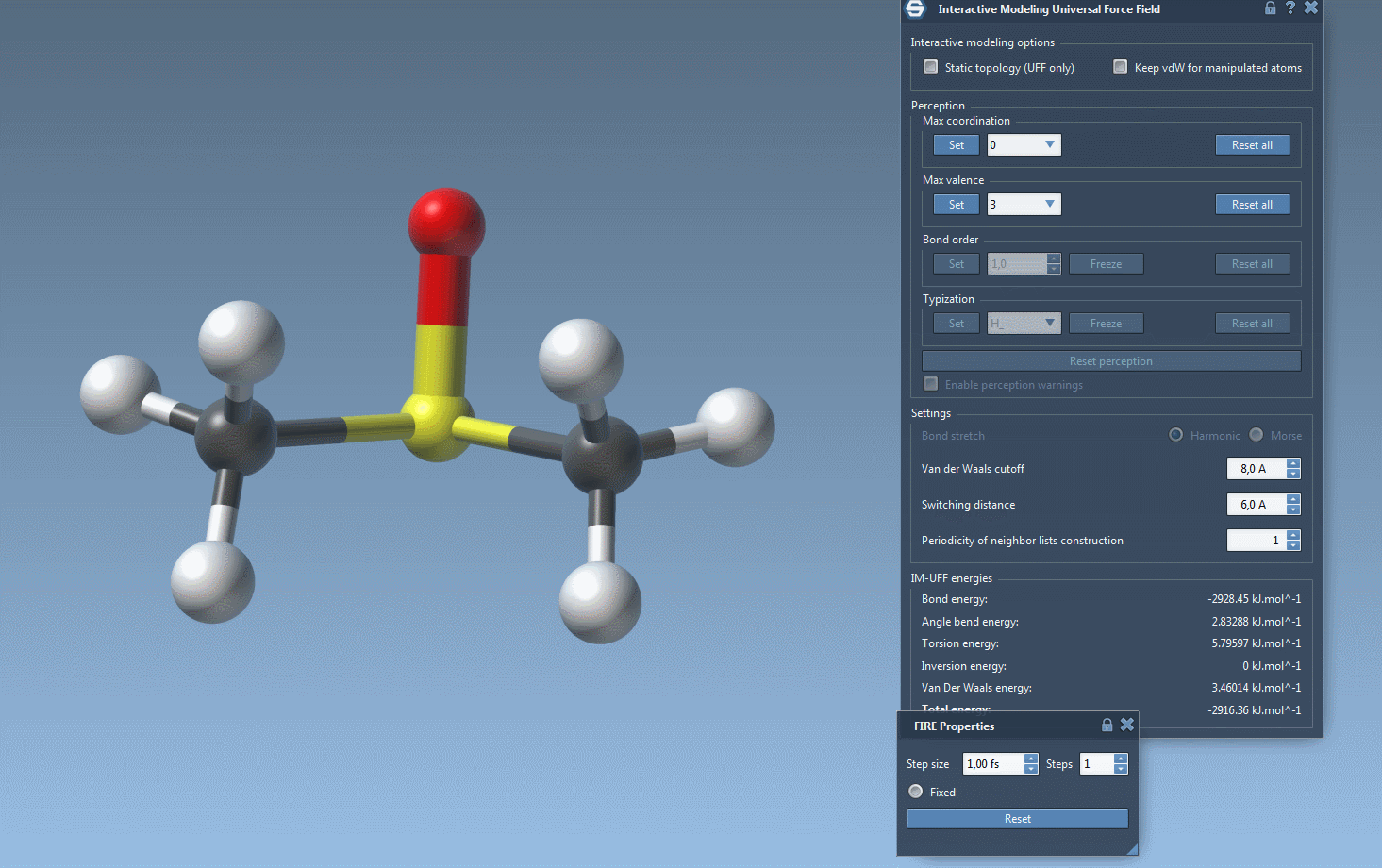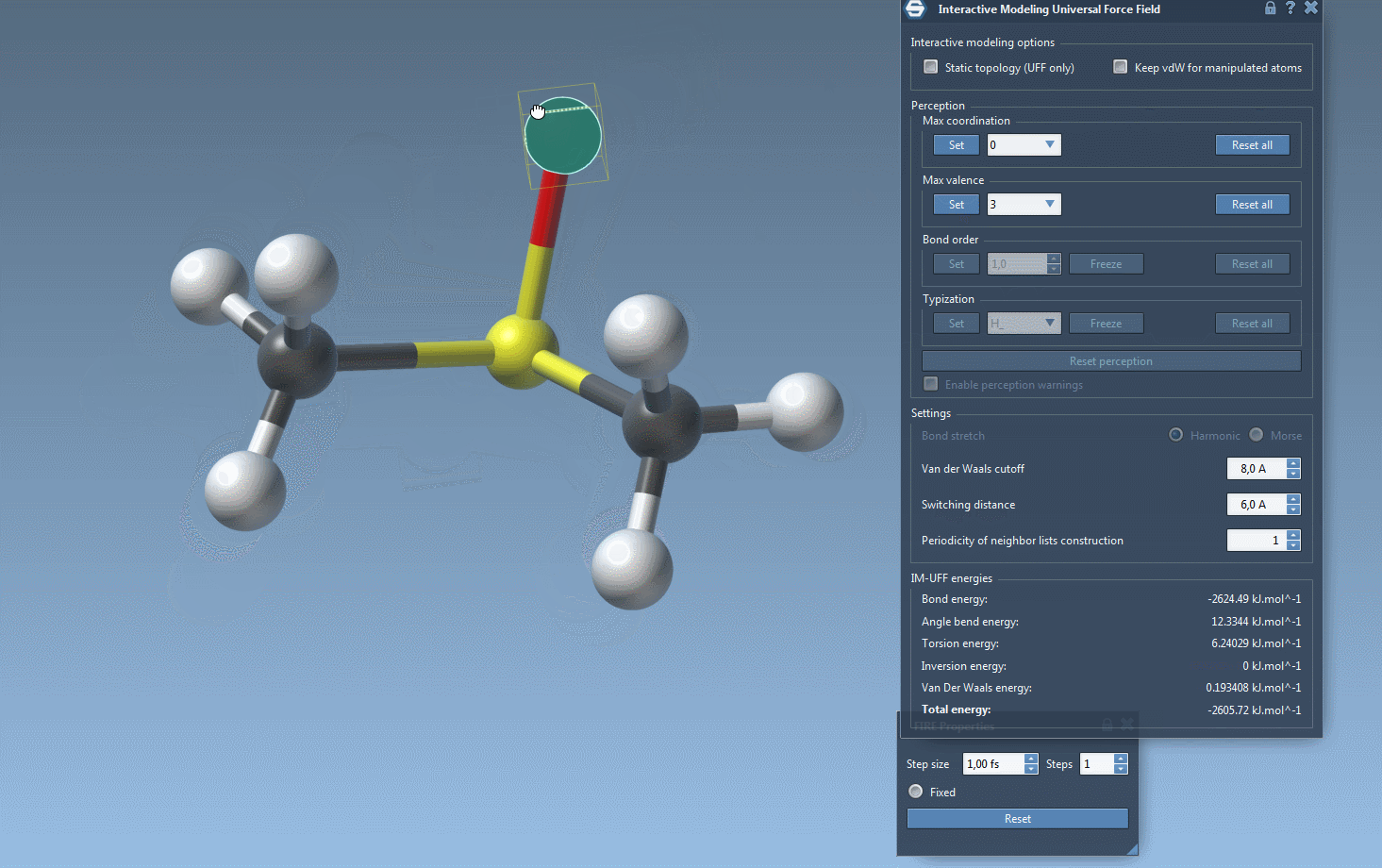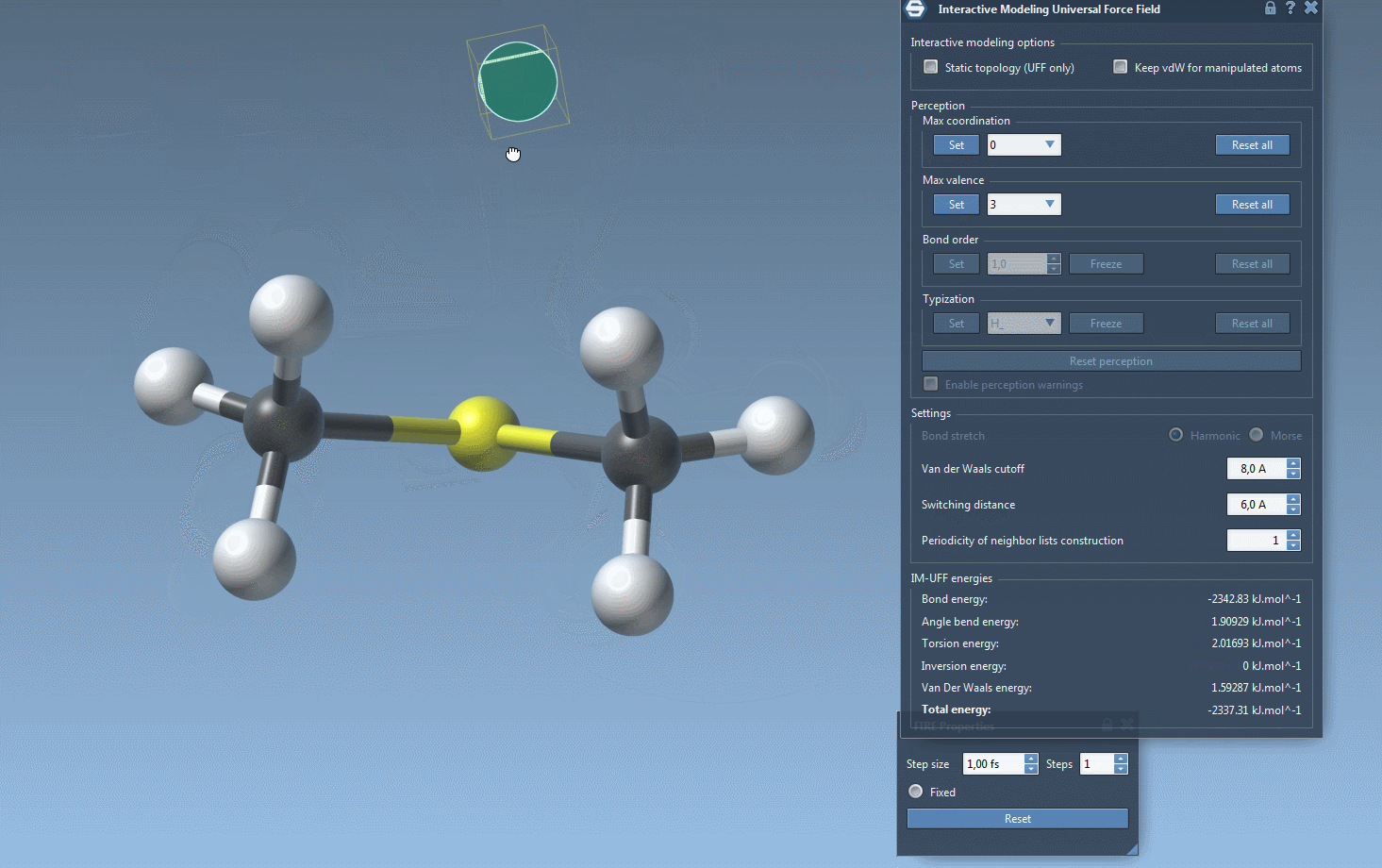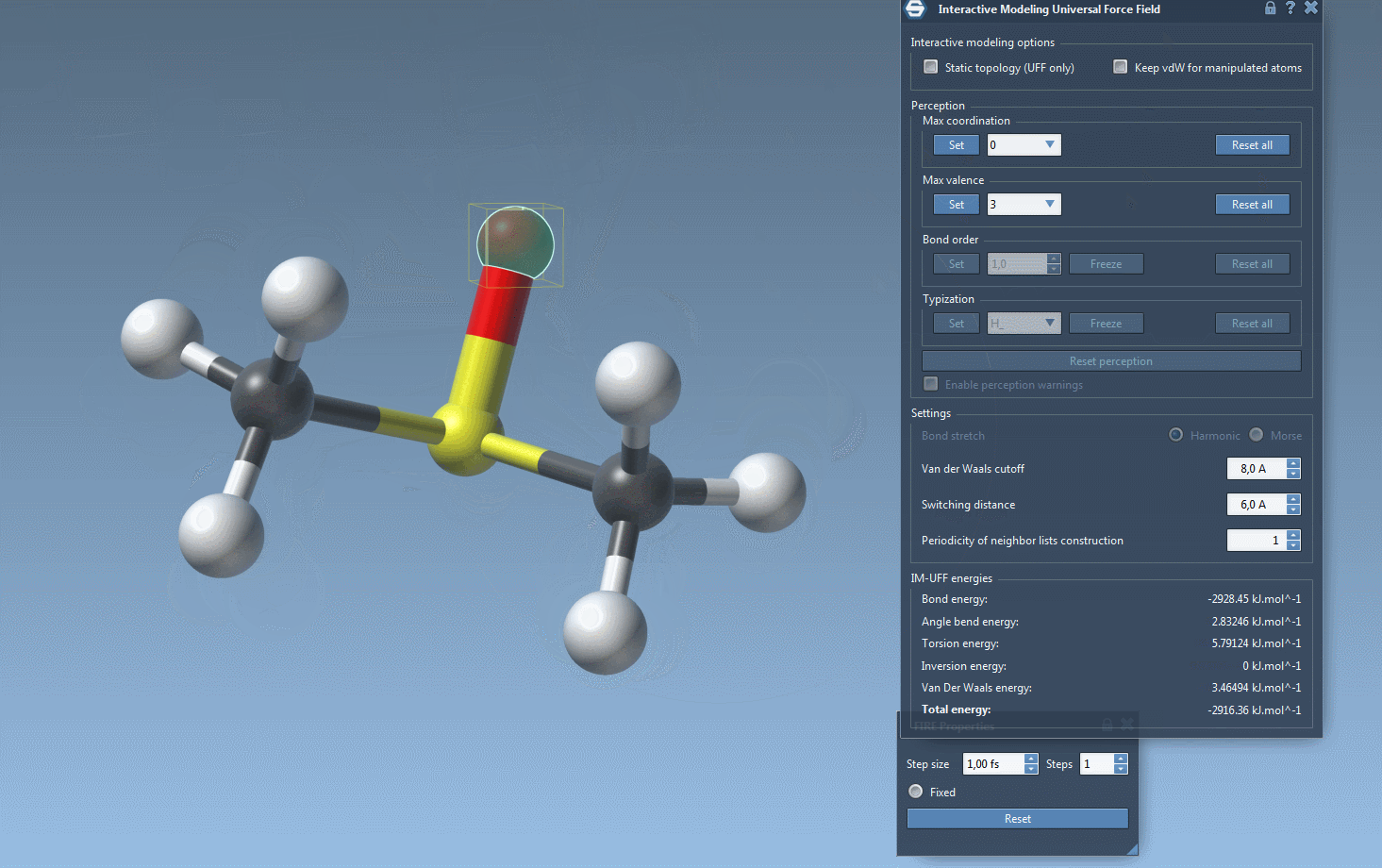
Watch topology transform dynamically as you drag atoms with the mouse. The model smoothly retains bonds for small movements but adapts to new configurations — breaking or forming bonds when atoms are drastically moved.

Customization Options for Advanced Users
IM-UFF also supports advanced customization, giving users control over parameters like:
- Van der Waals cutoff distance
- Switching distances for van der Waals interactions
- Periodic updates for the neighbor list construction
Additionally, the extension includes automatic typization of molecular systems. For users requiring deeper control over molecular rules, such as maximum coordination or valence settings, adjustments can be made to guide simulations more precisely. However, when running IM-UFF with topology changes enabled, bond orders and typizations naturally adopt continuous values for a seamless simulation experience.
Ready to Enhance Your Molecular Modeling?
Using IM-UFF can significantly improve interactivity and flexibility in your molecular design process by bridging traditional force field constraints with modern dynamic modeling tools. For more details and step-by-step instructions, visit the official documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON now at https://www.samson-connect.net.