





Molecular modeling often involves navigating the complexities of atomic interactions, bond formation, and structural modifications. A recurring challenge for modelers is the ability to perform these tasks seamlessly while visualizing and interacting with the molecular system in real-time. This is where the Interactive Modeling Universal Force Field (IM-UFF) extension in the SAMSON platform stands out as a solution.
The IM-UFF extension extends the familiar Universal Force Field (UFF) by allowing users to handle topological changes interactively. Whether you’re breaking or creating bonds, altering bond orders, or changing atom typizations, IM-UFF facilitates these operations dynamically and smoothly. This makes it ideal for constructing and editing molecular systems while being guided by physically-based inter-atomic forces.
Making Molecular Manipulations Intuitive
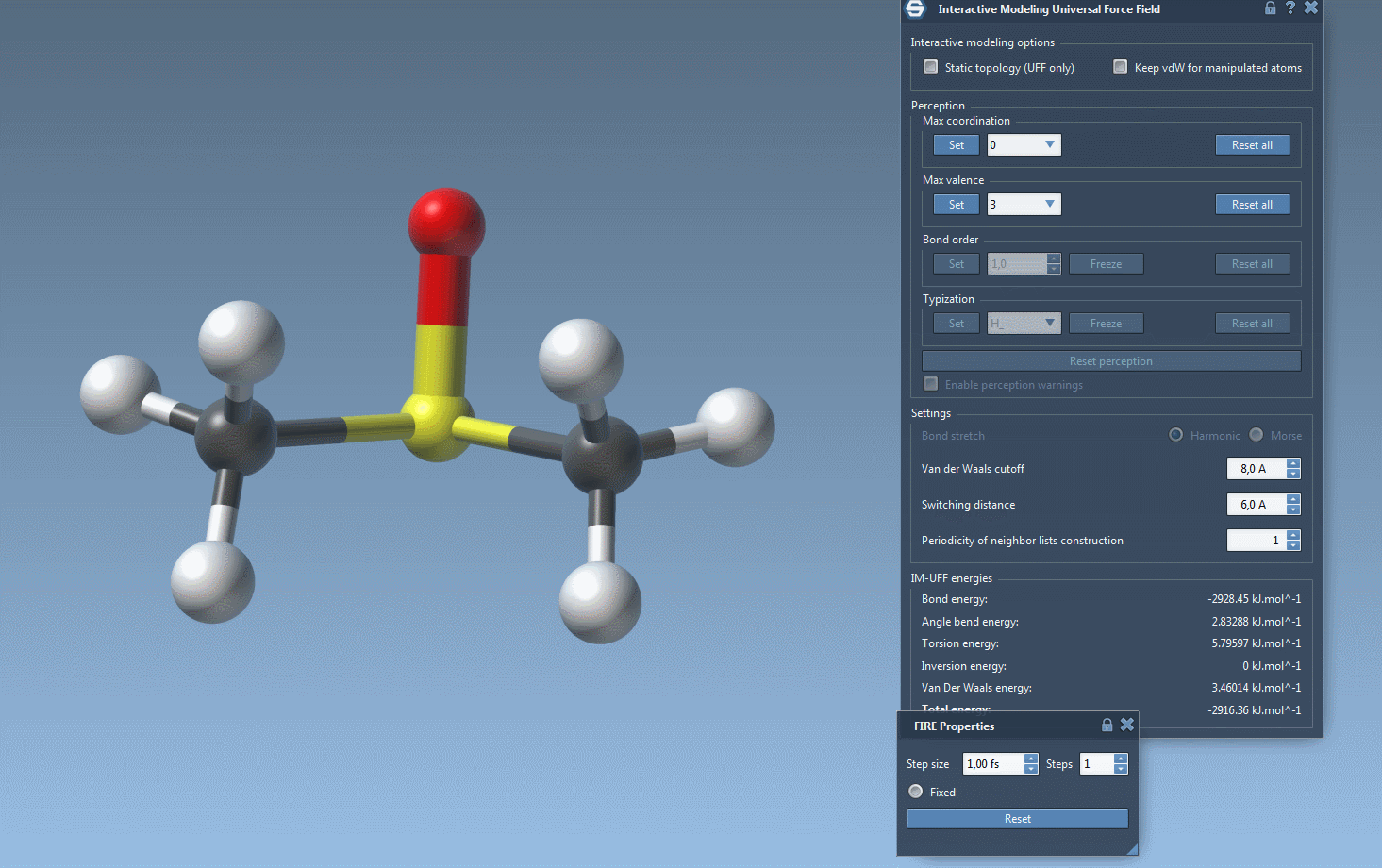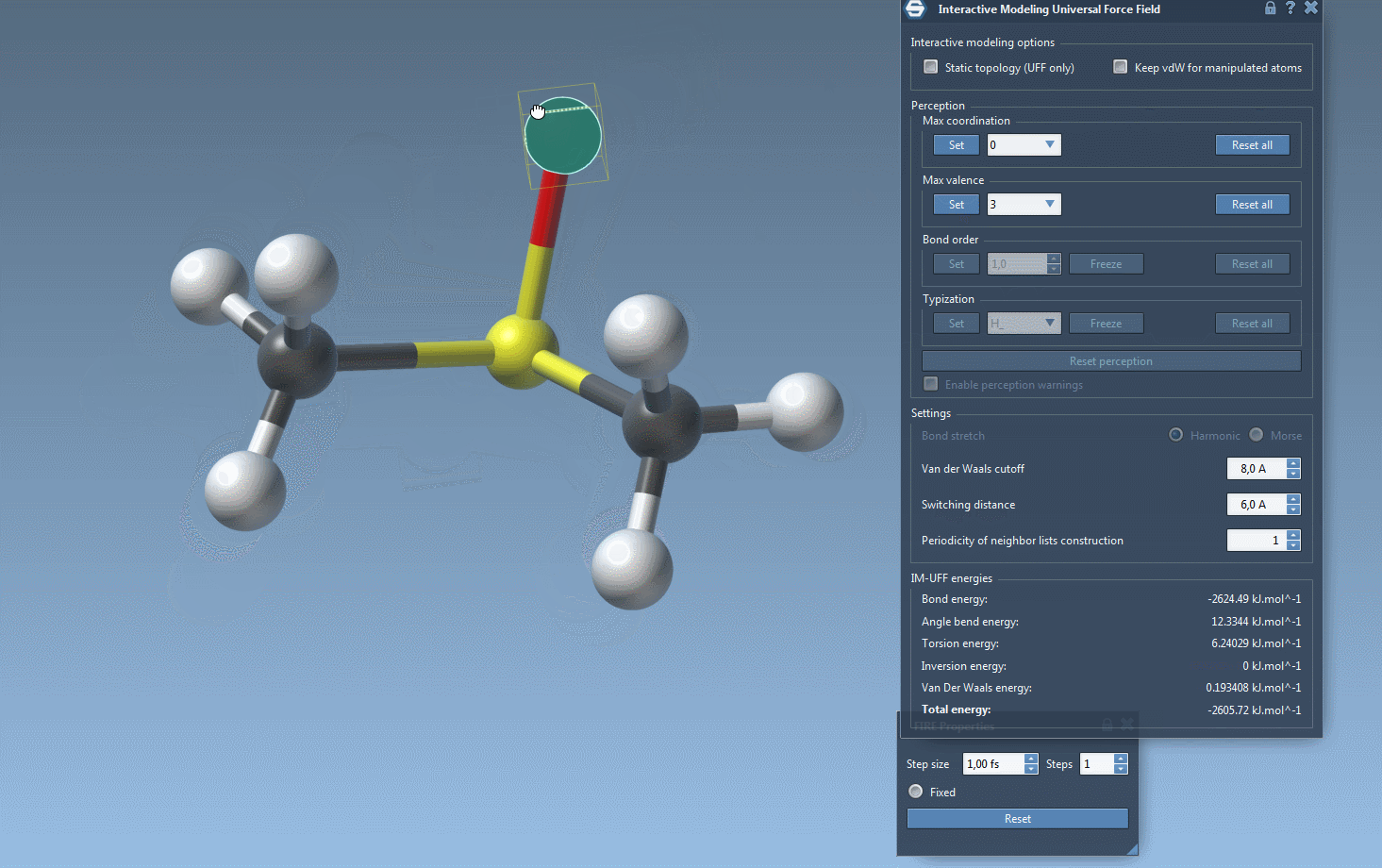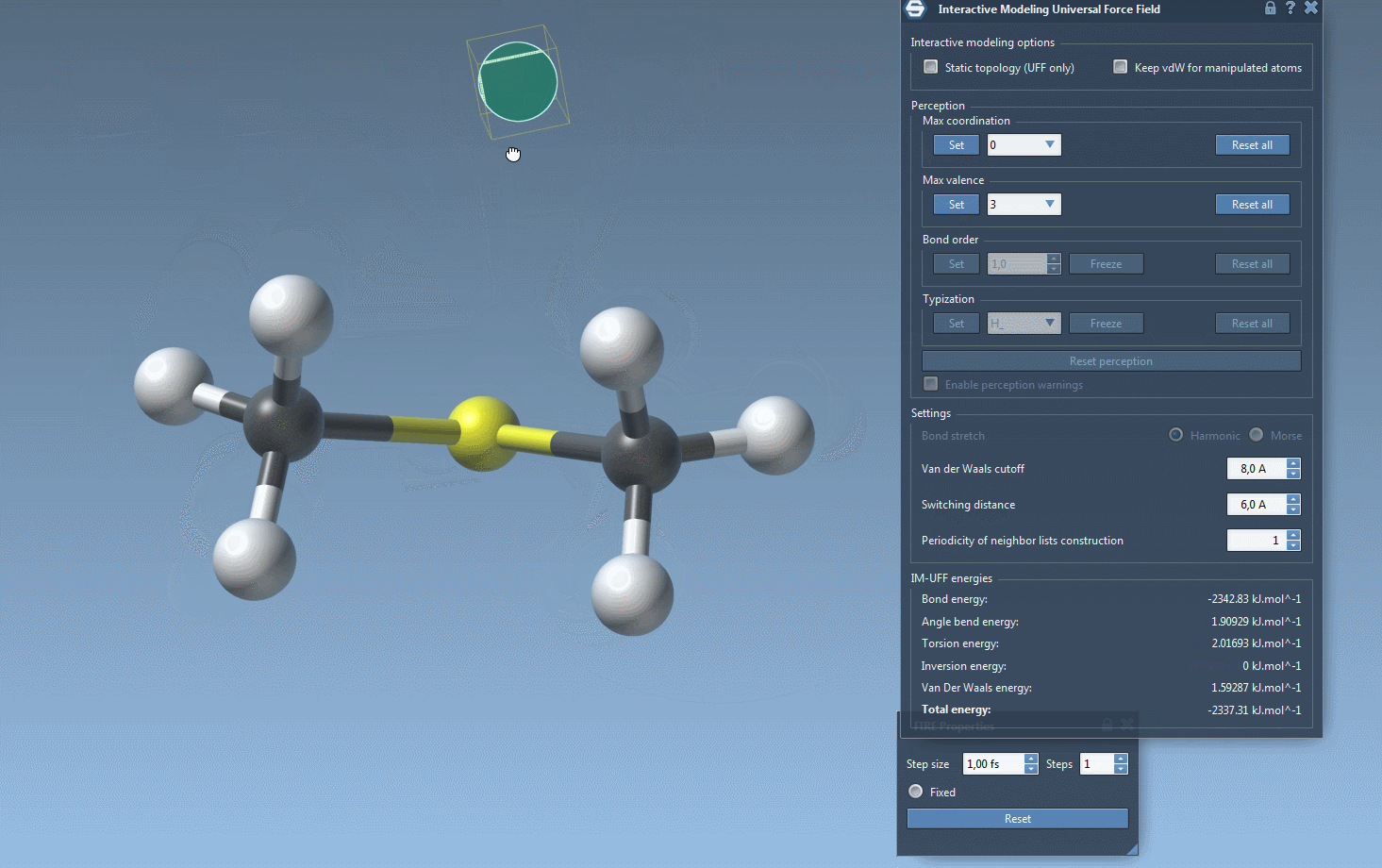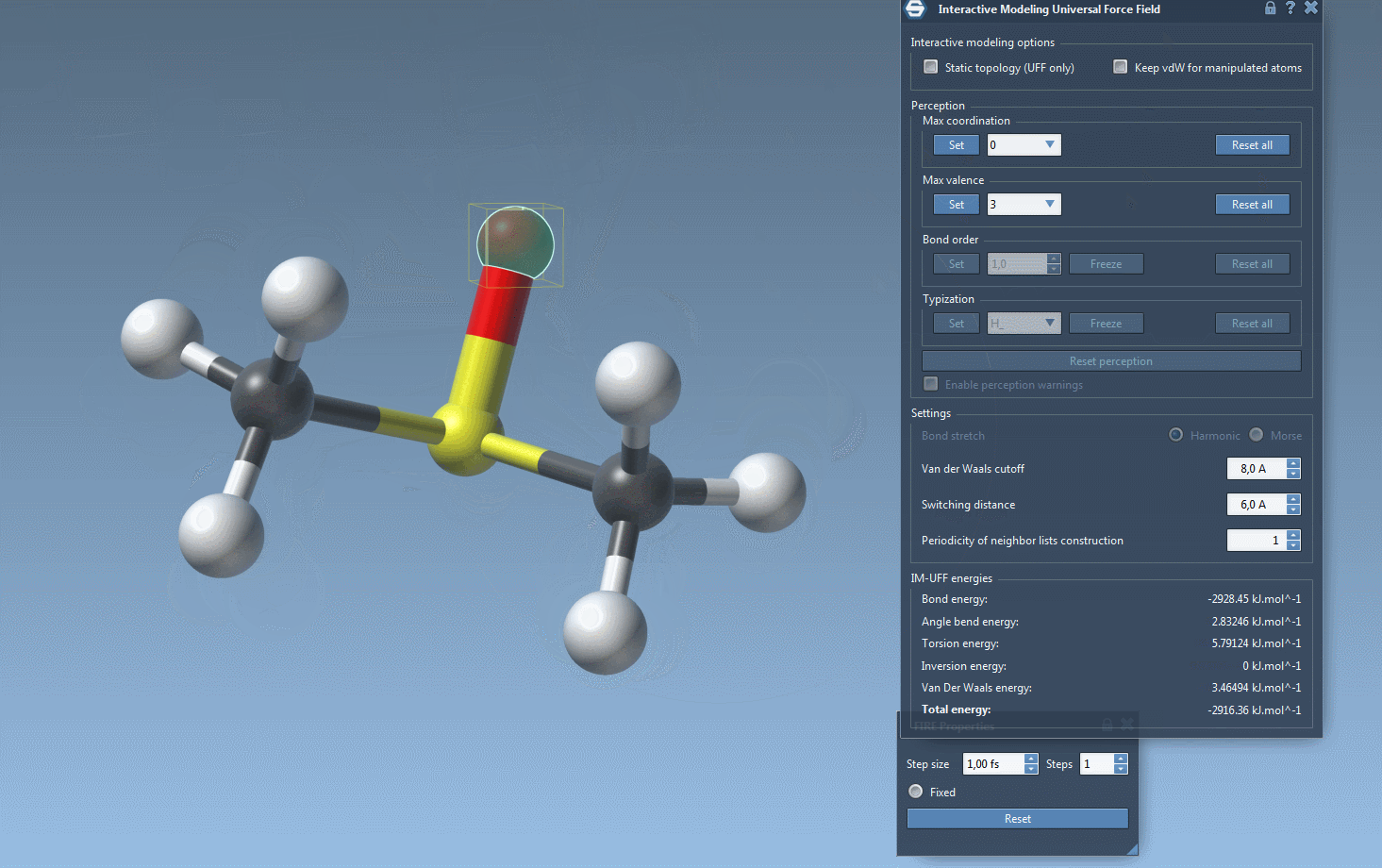
One of the standout features of IM-UFF is its ability to handle topological changes effortlessly. Imagine moving atoms within your molecular structure. Slight displacements may adjust the system locally while preserving the topology, but if you move an atom far enough, IM-UFF dynamically breaks bonds to its neighbors. Conversely, atoms brought closer will form new bonds, enabling the creation of updated topologies in real-time. This feature is a game changer for molecular modelers who need to iterate quickly and intuitively without cumbersome recalculations.

Interactive Options That Matter
IM-UFF introduces two crucial options in its parameter window to further enhance the modeling experience:
- Static topology (UFF only): This allows users to switch between standard UFF and IM-UFF, providing flexibility in maintaining or modifying molecular topologies. Notably, there’s a subtle difference in how bond energy is treated between the two modes, ensuring thermal stability across simulations.
- Keep vdW for manipulated: This setting comes into play when using IM-UFF. When enabled, it considers all van der Waals (vdW) interactions, while disabling it allows easier manipulation of atoms. For instance, this is particularly helpful when connecting manipulated atoms to others without interference from repulsive vdW forces.
Customizing IM-UFF for Your Needs
Customization within IM-UFF ensures a tailored experience for molecular modelers. You can set parameters like van der Waals cutoff distances and switching distances for neighbor list construction in both UFF and IM-UFF modes. Notably, when IM-UFF is running with dynamic topology, properties such as maximum coordination and valence become adjustable, while bond orders and atom typizations are calculated on the fly to allow smooth transitions.
Additionally, IM-UFF supports dynamic updates to your molecular system, including the removal or addition of atoms. Typizations are automatically recalculated, making it easier to iteratively refine your model without manual overhead.
Get Started with IM-UFF
Setting up an IM-UFF simulation in SAMSON is straightforward. After opening a molecular system, add a simulator through Edit > Simulate > Add simulator, and select “Interactive Modeling Universal Force Field” from the list. Choose a state updater like FIRE, and you’re ready to go. IM-UFF’s intuitive parameter window will help you fine-tune the simulation as needed.
Watching how IM-UFF adjusts topology as you interact with your molecular system in real-time can deepen your understanding of molecular mechanics and facilitate faster, insightful modeling workflows.
To dive deeper into the details and unlock the power of IM-UFF, visit the official tutorial page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON today at SAMSON Connect.