





For molecular modelers striving to improve the accuracy and realism of protein-ligand docking, accounting for ligand flexibility is crucial. Flexibility allows the exploration of different conformations and interactions within the binding site, making docking outcomes more reliable. However, controlling ligand flexibility and determining which bonds should rotate can be a complex process. This blog post provides a step-by-step guide on how to effectively manage ligand flexibility using the AutoDock Vina Extended SAMSON Extension.
Why Ligand Flexibility Matters
Proteins and ligands constantly adapt their structures to maximize interactions. With ligand flexibility enabled, rotatable bonds allow ligands to conform better to the binding site, simulating realistic conditions. Properly managing which bonds are rotatable (or not) can have a significant impact on docking efficiency and accuracy.
Setting Up Rotatable Bonds
Once you have set your ligand in SAMSON for docking, the AutoDock Vina Extended extension provides tools to visualize and manage rotatable bonds. Green cylinders on bonds in the Viewport represent rotatable bonds, while red cylinders indicate locked (non-rotatable) bonds.
To activate or deactivate a rotatable bond, click on the cylinders: green means the bond can rotate, and red means the bond is locked. If you want to zoom in on the ligand details while managing these bonds, select the ligand in the Document view and press Shift + Space to focus.
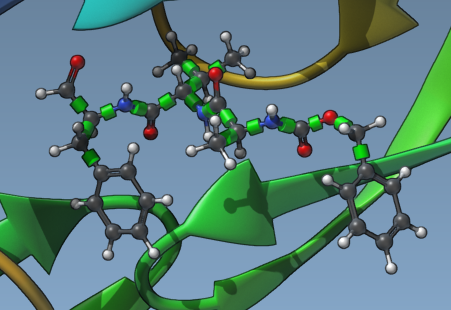
Locking Specific Bonds
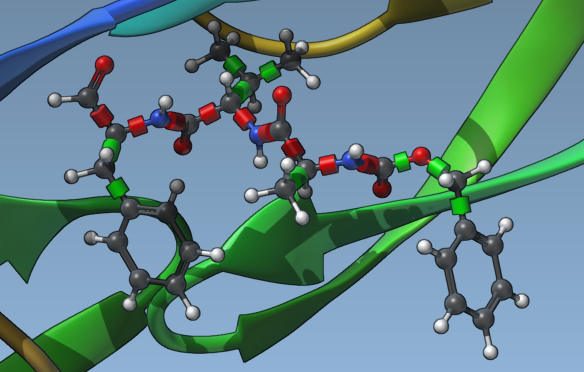
The tool also allows you to lock specific types of bonds across the ligand. To configure locked bonds:
- Click on Locked bonds settings.
- Check the bond types you want to lock (e.g., double bonds, aromatic bonds).
- Activate the Lock specific ligand bonds option.
This approach ensures that critical structural features remain stable during docking while allowing exploration of other parts of the molecule. Below is an example of a ligand with specific bonds locked:
Minimizing Ligands Before Docking
Sometimes, ligands in your library may not be in their optimal 3D conformations. To rectify this, you can minimize ligands directly using AutoDock Vina Extended:
- Check the Minimize option in the docking interface.
- Choose a minimization preset that defines the stopping criteria and maximum number of steps.
- Ensure that the Add missing hydrogens option is selected if hydrogens are not already present.
Proper minimization enhances ligand conformations, improving docking results. A well-prepared ligand often reduces computation time during docking simulations.

Balancing Realism and Computation Time
While enabling rotatable bonds leads to more realistic docking scenarios, it also increases computation time. By selectively locking specific bonds or categories and carefully pre-minimizing ligands, you can strike a balance between realism and computational efficiency.
Learn More
For more comprehensive instructions, including how to set up your docking project and analyze results, visit the original documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON at https://www.samson-connect.net.