





Protein-ligand docking can be an intricate process, especially when aiming for realistic simulations. One significant challenge molecular modelers often face is configuring rotatable bonds in ligands to strike a balance between computational efficiency and biologically accurate docking results. This post dives into how to set up and adjust rotatable bonds using the AutoDock Vina Extended extension in SAMSON, a step-by-step process that can help optimize your docking workflows.
Why Adjust Rotatable Bonds?
In molecular docking, ligands often contain bonds that can rotate freely, allowing the ligand to explore different conformational states during docking. While enabling rotatable bonds introduces flexibility and provides more realistic docking results, it can also increase computational time significantly. Thus, identifying which bonds to allow or restrict plays a crucial role in efficient and effective docking workflows.
Setting Up Rotatable Bonds
To start, ensure you have your ligand ready and loaded into SAMSON. Select the AutoDock Vina Extended app from the Home > Apps > Biology menu, or use the Find everything… search box (Shift + E). Then, proceed as follows:
1. Initial Binding Configuration
Under the “Set ligand” section, choose Single ligand. Next, select your ligand in the Document view and click the Set button. Once set, you will see green cylinders in the Viewport, representing rotatable bond controllers superimposed on the ligand’s bonds.
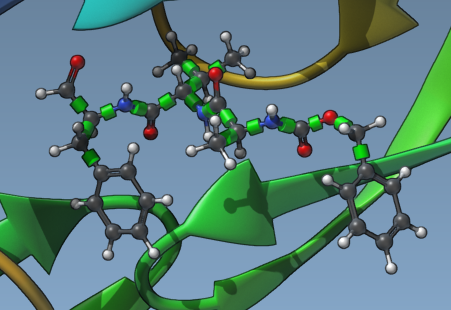
2. Adjusting Rotatable Bonds
Clicking on a green cylinder toggles the bond state between rotatable and not rotatable. Red cylinders indicate locked bonds, while green ones indicate active flexibility. Use this functionality to lock bonds that don’t require rotation, optimizing computational workload.
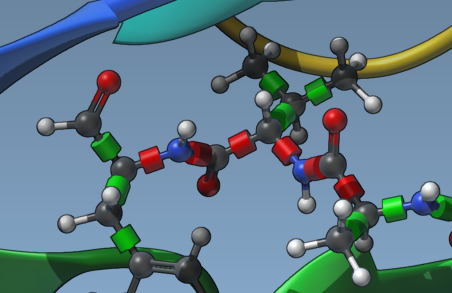
3. Locking Specific Bond Types
If you want to lock specific types of bonds across all ligands systematically, click Locked bonds settings. This option allows you to specify bond types (such as amide bonds) that should remain locked, ensuring uniform parameter application across simulations. Check the Lock specific ligand bonds box afterward.
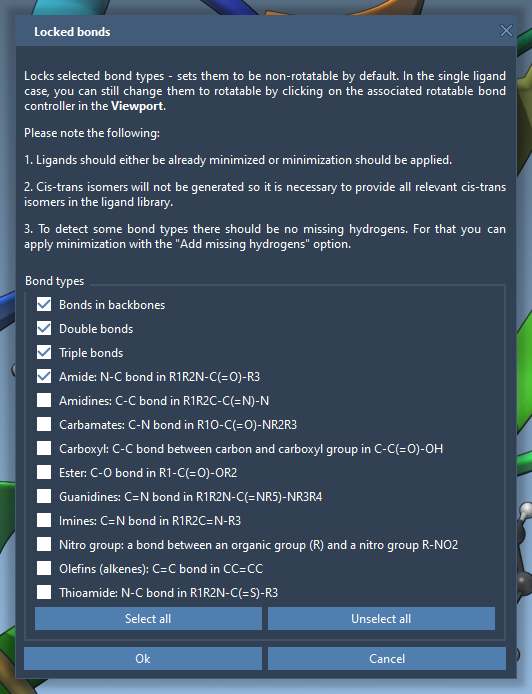
4. Ligand Library Flexibility
If docking a library of ligands, note that all bonds are considered rotatable by default. However, you can apply the same locking principles to the entire library using Locked bonds settings. This ensures consistency and effectively reduces runtime for large-scale projects.
When to Minimize Ligands?
Minimizing ligands is important if your ligands are not in their most stable conformations, as this can impact docking accuracy. Before initiating docking, use the Minimize option to reconfigure ligands into optimal geometries. Adding missing hydrogens is also crucial for detecting polar interactions such as hydrogen bonds.

Improving Docking Performance
Configuring rotatable bonds thoughtfully not only aligns with the biological relevance of the docking but also allows you to balance realism and computation time effectively. Start practicing by toggling bonds interactively or applying systematic lock rules to ligand libraries—both approaches significantly enhance your results.
For a complete guide and additional details, visit the full documentation at SAMSON’s tutorial page.
*Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON at www.samson-connect.net.