





Simulating molecular systems can be complex, especially when you're looking for an integrative force field that handles a wide range of simulations effectively. For molecular modelers, the Universal Force Field (UFF) in SAMSON offers an automated and flexible way to calculate molecular interactions while saving time on manual adjustments.
In this guide, we'll walk you through setting up and running an UFF simulation within SAMSON, and explore how this tool simplifies interaction modeling for molecules. If you've ever struggled with managing atom types, bond orders, and energy calculations for large molecular systems, learning this feature is a must.
Setting up UFF in SAMSON Made Easy
To start using the UFF, follow these straightforward steps:
- Open your molecular system document.
- Add a simulator by navigating to
Edit > Simulate > Add simulatoror using this shortcut: Ctrl + Shift + M on Windows / Cmd + Shift + M on macOS. - Select Universal Force Field from the interaction model list.
- Pick your preferred state updater for the simulation, like FIRE.
- Click the OK button. This step opens the UFF setup window.
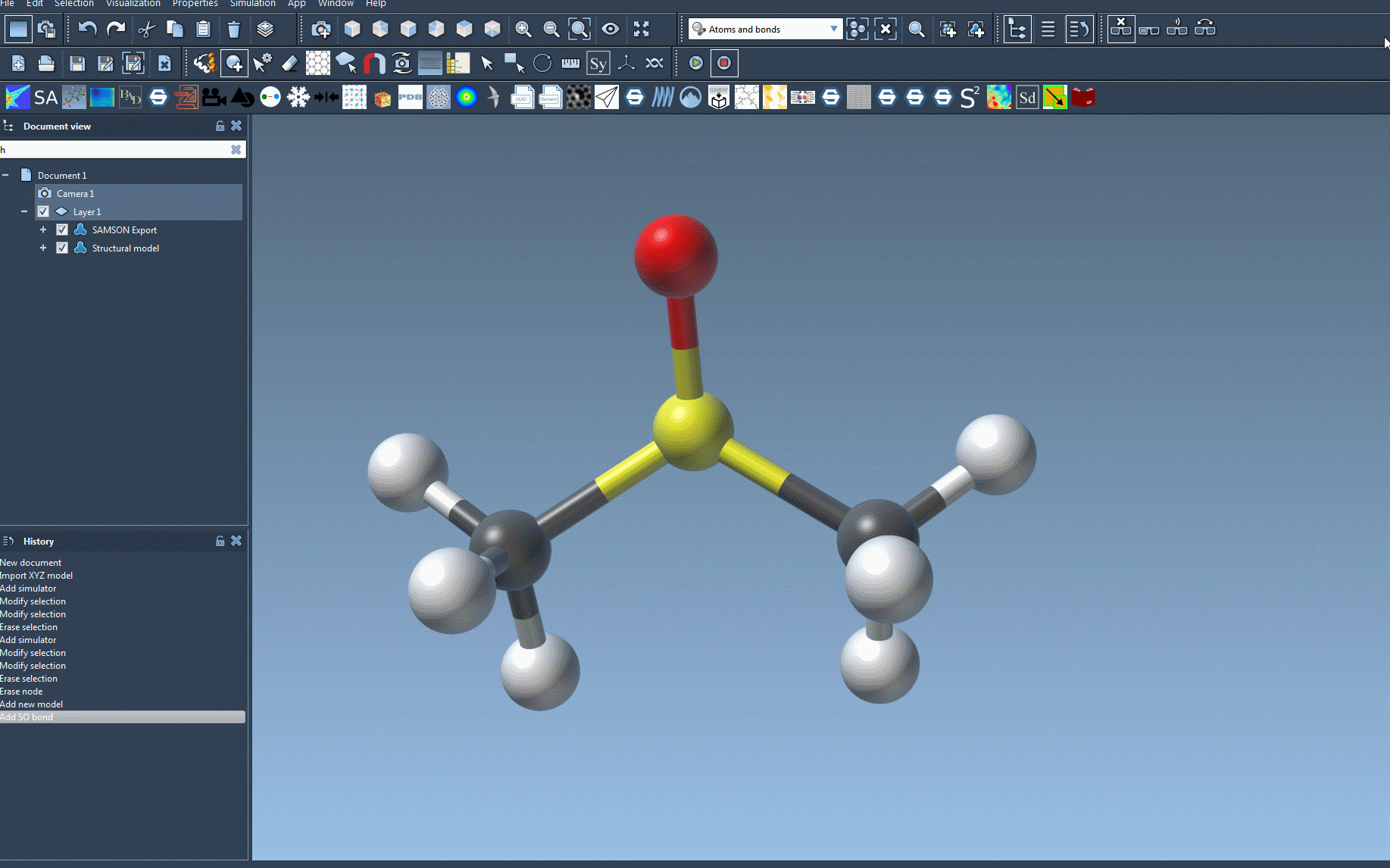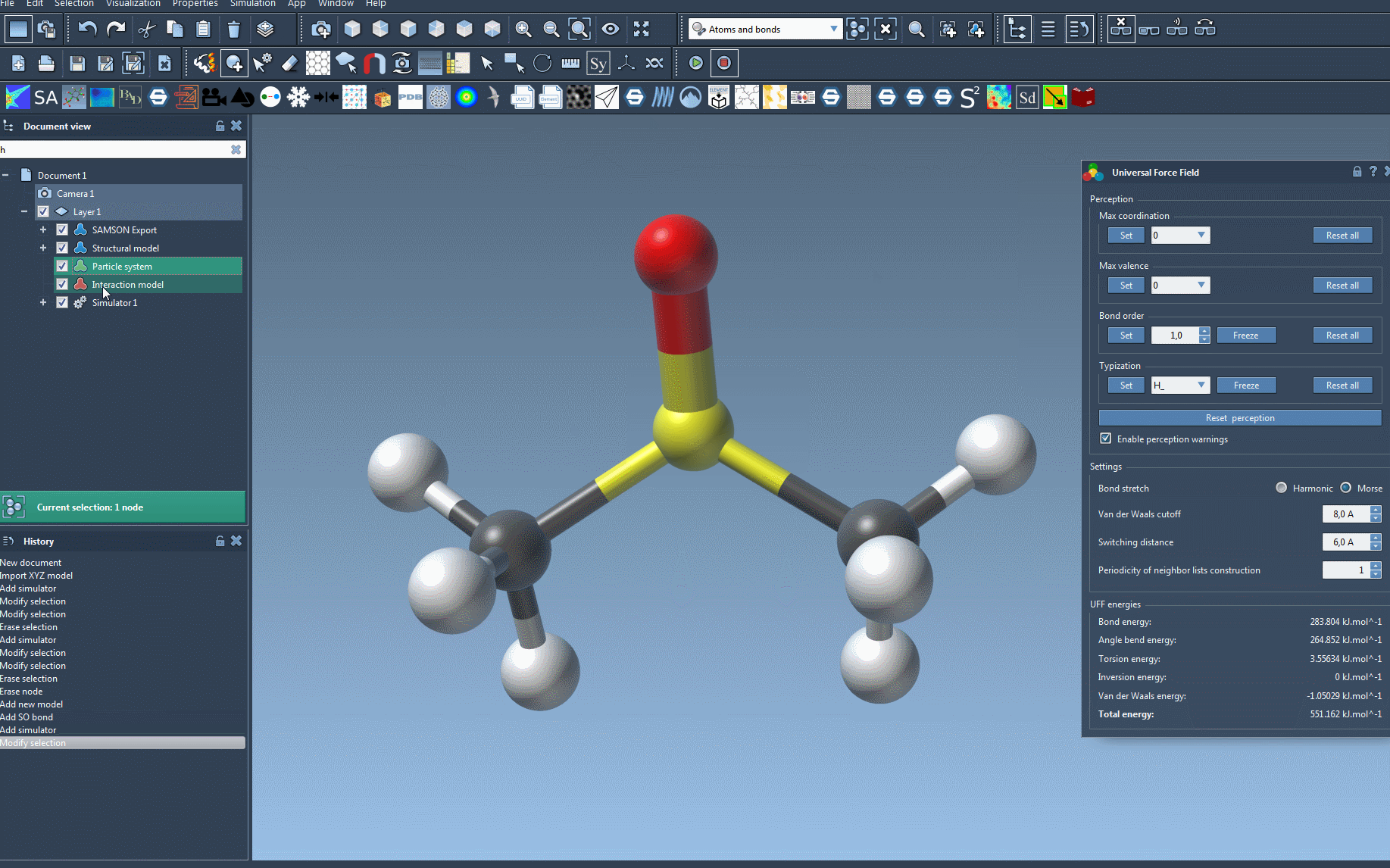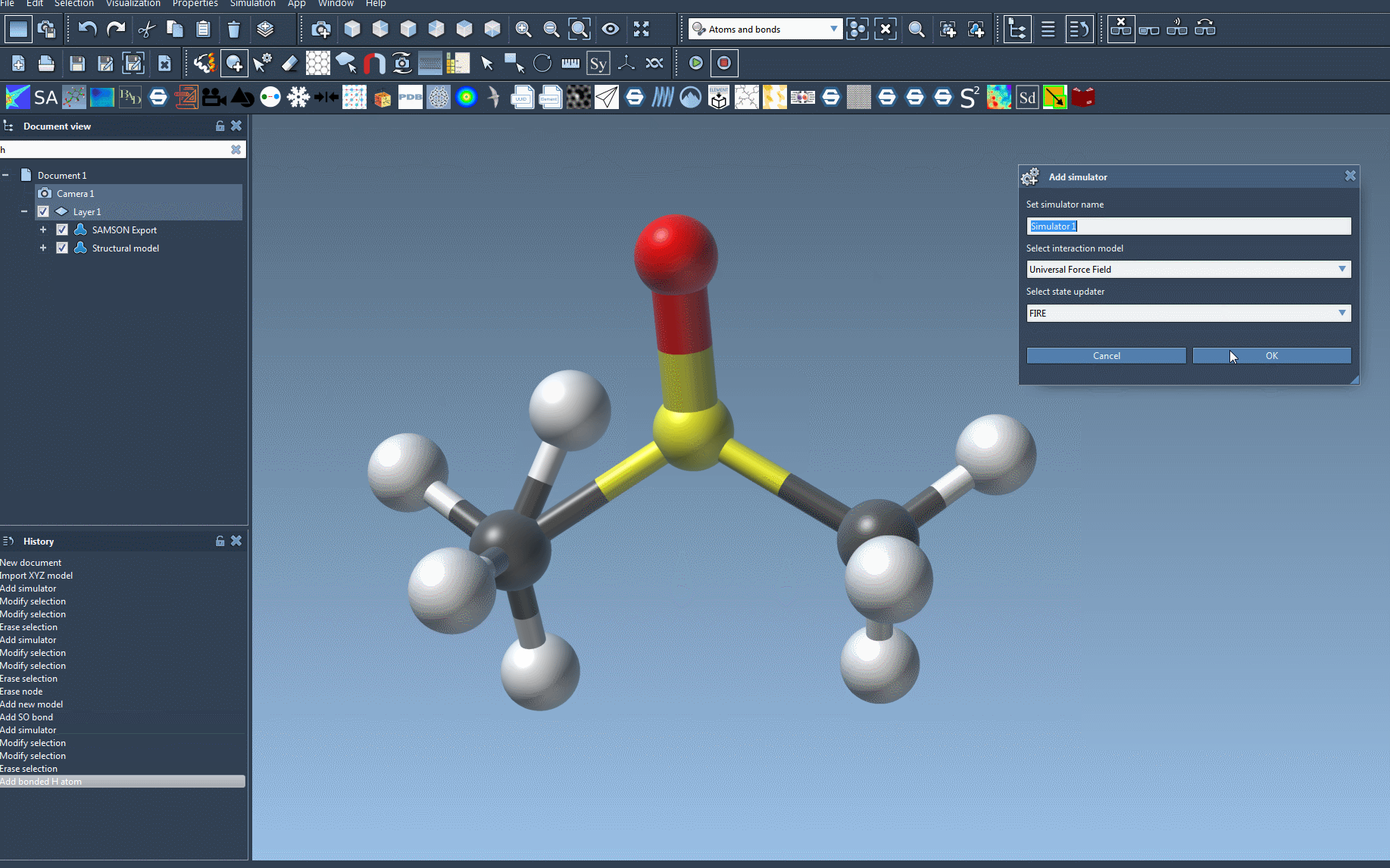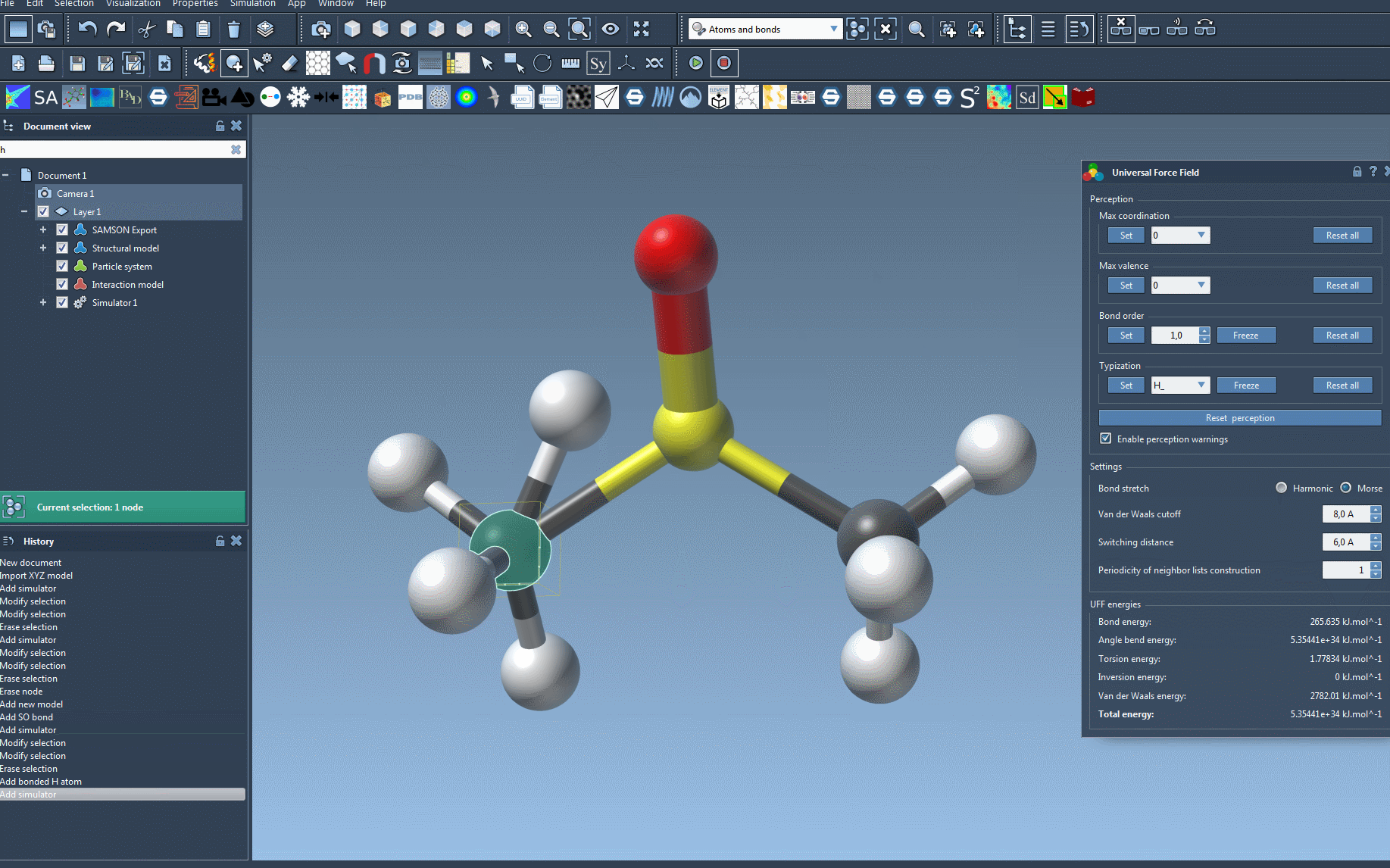
In the setup window, you can decide whether to use existing bonds (if they've been predetermined) or let UFF automatically compute molecular bonds, bond orders, and atom types. After confirming your preferences with OK, the UFF module initializes your molecular system by:
- Computing covalent bonds (if Use existing bonds was not checked).
- Determining bond orders and atom types.
If there are any inconsistencies detected during initialization, SAMSON will notify you with a warning message. Once setup is complete, you're ready to move to the next step: running the simulation!
Running the UFF Simulation
Once your system is set up, running the UFF simulation is simple. Just press Edit > Simulate > Start. While the simulation is running, you'll see live feedback on the energy of each UFF term alongside the total simulation energy.
One of the most useful features during the simulation is the ability to interactively move atoms. As you adjust the position of specific atoms, you'll see how the UFF model responds dynamically by rearranging the positions of other atoms. For example, this can help you understand how energy terms like van der Waals interactions influence molecular geometry in real-time.
Customizing the Interaction Parameters
If you want even more control over the simulation, UFF offers a range of customizable parameters:
- Bond stretch interactions: Switch between Harmonic and Morse types.
- Van der Waals (vdW) cutoff distance and switching distance: Define how vdW interactions are computed across distances.
- Neighbor list periodicity: Choose how often neighbor lists should be recomputed at each timestep.
These options allow you to tailor simulations to match your specific molecular modeling needs. Experimenting with them can provide insights into molecular dynamics that are harder to observe with default settings.
Why Use UFF in SAMSON?
What makes UFF particularly valuable is its comprehensive automation. It minimizes the need for manual input by automatically perceiving bonds, bond orders, and atom types based on molecular structure. This feature significantly reduces setup time, especially for large and complex systems.
Moreover, UFF's flexibility allows advanced users to customize atom typization and bond orders, enabling deeper and more precise control over simulations. Whether you're testing new molecular designs or exploring dynamic behaviors, the UFF module in SAMSON is a solid solution to streamline your modeling processes.
Ready to dive deeper? Learn more through the official documentation page for detailed explanations, examples, and references.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at SAMSON-Connect.