





One of the key challenges in molecular docking is achieving an optimal balance between computation time and docking accuracy. Ligand flexibility, denoted by the configuration of rotatable bonds, plays a crucial role in the docking process. With the AutoDock Vina Extended SAMSON Extension, configuring rotatable bonds effectively can lead to more realistic receptor-ligand interactions while maintaining time efficiency. This blog post guides you step-by-step on how to maximize the benefits of this feature in ligand docking workflows.
Why Rotatable Bonds Matter
Rotatable bonds offer the flexibility needed for a ligand to adapt to a protein’s binding pocket, improving docking accuracy. However, they also introduce additional computational complexity, which can extend the docking time significantly, especially for libraries with numerous ligands.
Step-by-Step Guide to Configuring Rotatable Bonds
Here’s how you can configure rotatable bonds in the AutoDock Vina Extended extension for SAMSON:
1. Setting Up the Ligand
Begin by selecting the ligand that you wish to dock. Ensure the Single ligand option is checked under Set ligand. In the Document view, select your ligand—for example, “2AZ8-IA”—and click the Set button. You will now see rotatable bond controllers, represented by green cylinders, in the Viewport covering the bonds of the ligand.
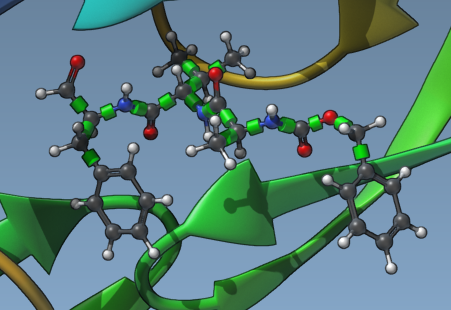
2. Activate or Deactivate Rotatable Bonds
Click on the green cylinders to toggle between rotatable (green) and locked (red) states. This action allows you to selectively activate or deactivate bonds based on the ligand’s chemical features and intended docking scenario. For example, locking rigid molecular features ensures computational effort is focused on more relevant degrees of freedom.
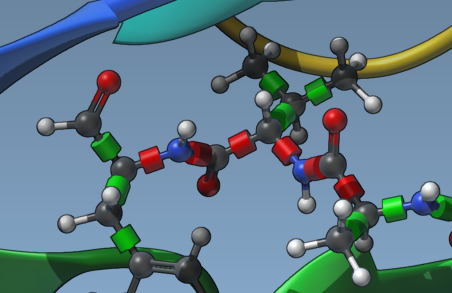
3. Lock Specific Bond Types
To globally apply restrictions on certain bond types, click the Locked bonds settings button. A dialog opens where you can specify bond types to be locked (e.g., double or aromatic bonds). Once configured, check the Lock specific ligand bonds option to apply this setting, as shown below:
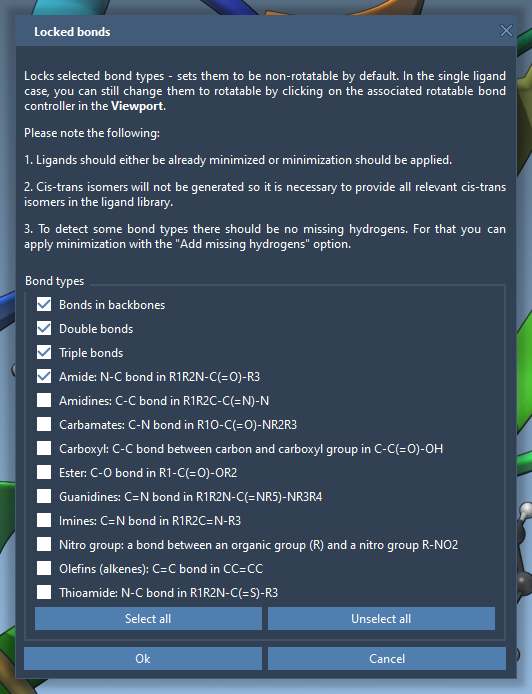
This feature ensures systematic control over ligand flexibility for more robust docking results.
4. Minimize Ligands When Necessary
Before initiating docking, it’s crucial to ensure your ligands are in their lowest energy conformations, especially for 2D ligands. Use the Minimization option to refine 3D configurations while adding any missing hydrogens required for docking. This process eliminates biases from non-optimal starting geometries and improves docking reliability.

Striking the Right Balance
While enabling rotatable bonds often enhances docking realism, consider docking time and computational capacity. For high-throughput workflows with large ligand libraries, selectively disabling non-essential rotatable bonds offers a strategic compromise. Similarly, evaluating chemical knowledge of your ligand can help pre-lock bonds that are unlikely to exhibit flexibility in binding interactions.
Ready to Enhance Your Docking Workflow?
Effectively managing rotatable bonds with the AutoDock Vina Extended extension in SAMSON can transform your molecular docking workflows, delivering more biologically accurate results while optimizing computing resources. This fine-tuned flexibility ensures you maintain control over docking parameters tailored to your specific research needs.
For more details and advanced options, visit the full documentation page here.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON at https://www.samson-connect.net.