





For molecular modelers, finding realistic transition paths between conformations can be a challenging task. These paths are critical for understanding dynamic processes like ligand binding or conformational changes in proteins. However, generating an initial path is only part of the job – refining it into a more physically meaningful transition can make your results significantly more valuable. Enter the Parallel Nudged Elastic Band (P-NEB) method in SAMSON. This tutorial will show you how P-NEB helps to optimize transition paths, step by step.
The Problem
When dealing with trajectories or pathways, the initial path often lacks physical realism or energy optimization. For example, in ligand-protein interactions, a simple linear interpolation between endpoints doesn’t necessarily reflect the true molecular movement or energy landscape. A rough, unoptimized path can impact the accuracy of simulations and free energy calculations.
What P-NEB Does
The P-NEB app in SAMSON tackles this pain by optimizing intermediate points (“images”) between initial and final states. Using a combination of spring forces and optimization algorithms, it distributes transitional states evenly while climbing potential energy landscapes to locate saddle points.
How to Use P-NEB for Path Refinement
To get started with P-NEB, follow these steps:
1. Load Sample Paths
You can quickly bring in sample documents available in SAMSON. For instance:
- A Zinc ligand unbinding trajectory: Download here.
- A protein-ligand complex (Lactose Permease and Thiodigalactosid): Download here.
This not only saves setup time but also ensures you work with realistic examples. Below is the interface for downloading sample projects:

2. Launch the P-NEB App
In your SAMSON workspace, navigate to Home > Apps > All > P-NEB, or simply search for “P-NEB” using the search field. The interface looks like this:
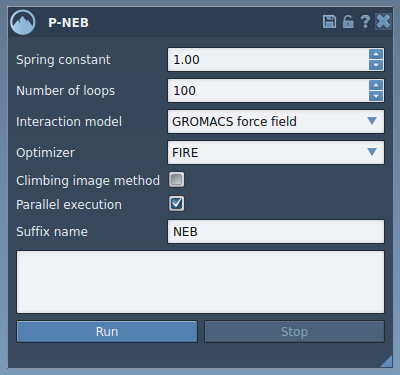
3. Adjust Key Settings
Before running the app, ensure the following settings are configured:
- Spring constant: Use 1.00 (default value).
- Number of loops: Input 100 for optimal refinement.
- Interaction model: Choose “Universal Force Field” for energy calculations.
- Optimizer: Select “FIRE” (Fast Inertial Relaxation Engine).
- Parallel execution: Check the box to enable multi-threaded computation.
4. Apply P-NEB
To refine a path, select it in the Document view, and click Run in the P-NEB app. The computation progress will be visible in the status bar:

Upon completion, a new, optimized path is generated and appears in the Document view for further inspection:
5. Analyze the Results
You can now examine the optimized path using tools like the Inspector. Double-clicking the path allows you to start/stop an animation of the transition, which is particularly helpful for visualizing molecular dynamics.
Tips and Notes
While P-NEB can also refine a set of conformations directly, it is generally more efficient to combine conformations into a single path beforehand using Conformation > Create path from conformations. This approach minimizes computation time and simplifies analysis.
Concluding Thoughts
The P-NEB app in SAMSON provides an accessible, powerful way to enhance molecular transition paths, making your simulations more accurate and credible. If you’d like to dive deeper into the nitty-gritty, visit the full tutorial documentation here.
SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON today at https://www.samson-connect.net.