





Identifying the minimum energy path between two molecular conformations is a fundamental task in molecular modeling. Whether you’re studying ligand unbinding, conformational rearrangements, or molecular switching, knowing how molecules move from one state to another—the so-called transition path—is key.
However, calculating transition paths isn’t always straightforward. Traditional optimization approaches may miss the saddle point or fail to preserve even spacing between intermediate states. This is where the Parallel Nudged Elastic Band (P-NEB) method in SAMSON can help. P-NEB refines a given path by applying spring forces and force-field-based energy minimization in parallel, improving both accuracy and computational speed.
What’s the problem?
Molecular modelers often use trajectory sampling tools like Molecular Dynamics (MD) or path generation methods such as linear interpolation to create approximate reaction pathways. But these can be inefficient or imprecise:
- Paths may not pass through transition states (saddle points).
- Conformations might not be spaced uniformly.
- Energy profiles can be noisy or unrealistic.
The P-NEB method can optimize these paths to identify more accurate transition mechanisms by refining submitted trajectories based on physics-based interaction models.
How to use P-NEB on an existing molecular path
In this tutorial, we’ll focus on how to apply the P-NEB approach to an existing molecular path in SAMSON.
Step 1: Load a sample model
From Home > Download in SAMSON, paste this document URL to load a sample ligand unbinding trajectory:
|
1 |
https://www.samson-connect.net/documents/39260103-9a48-49db-9488-5573d5f1e7b0 |
This model describes a Zinc ligand unbinding pathway and is ideal for testing.

Step 2: Launch the P-NEB App
Open the app from Home > Apps > All > P-NEB. The interface looks like this:
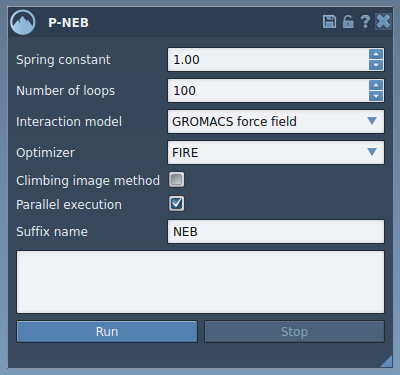
Step 3: Select and Optimize a Path
In SAMSON’s Document view, select the path node (trajectory).
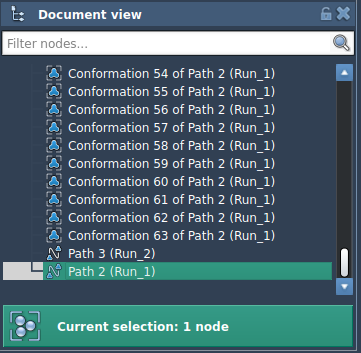
Set the following P-NEB parameters:
- Spring constant: 1.00
- Number of loops: 100
- Interaction model: Universal Force Field
- Optimizer: FIRE
- Parallel execution: Checked
- Suffix name: NEB
Then press Run. When prompted by the Universal Force Field (UFF) setup, choose to use existing bonds and click OK.

Step 4: Monitor and Inspect Results
You can follow the computation progress in the status bar:

Once the optimization is done, a new path appears in the Document view with “NEB” in its title.
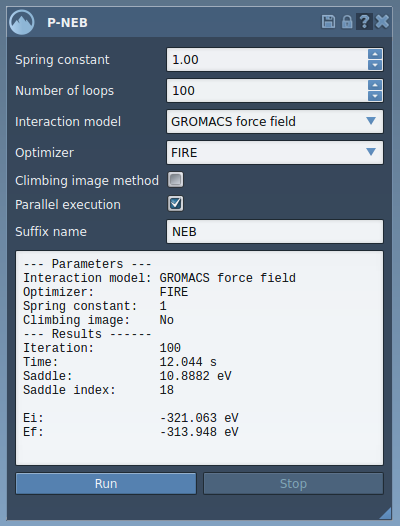
Select the new path and use the Inspector to analyze the positional and energetic outcomes. You can also double-click to animate the transition or use the right-click menu for additional controls.
Final remarks
Refining an approximate molecular transition pathway into a minimum energy route with P-NEB improves both realism and interpretability. If you already have a trajectory—whether generated manually or using another tool—P-NEB gives you a scientifically grounded way to optimize it without starting from scratch.
Learn more in the full tutorial at this link.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.