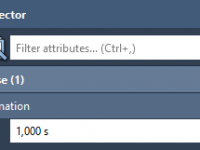
Master Static Views with the Hold Camera Animation in SAMSON
Understanding and Using SAMSON Apps Efficiently
Effortlessly Create Replicas for Coarse-Grained Modeling in SAMSON
If you are a molecular modeler aiming to create coarse-grained models for systems with multiple replicas, you’ve probably encountered challenges around preparing and organizing replicas of biomolecules. The errors stemming from improper numbering or overlapping structures can significantly disrupt workflows.…
Mastering Light Attributes in SAMSON’s Node Specification Language
Easily Generate Protein Transition Paths with ARAP Interpolation.
Streamline Your Molecular Design with Dark Mode in SAMSON.
Efficient Molecular Modeling with Segment Attribute Insights
Ensuring a Consistent View in Molecular Animations with ‘Hold Camera’
For molecular modelers and researchers, visual consistency is essential when creating animations. Whether you’re examining a protein’s folding process, highlighting molecular interactions, or presenting dynamic simulations, maintaining a steady point of view can make your presentations cohesive and professional. However,…