Understanding Structural Changes with RMSD Analysis in SAMSON
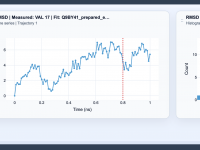
One frequent challenge in molecular modeling is distinguishing between overall rigid-body motion and meaningful internal structural changes. This distinction is crucial for making sense of conformational dynamics, binding events, and other molecular processes. The RMSD (Root Mean Square Deviation) analysis…
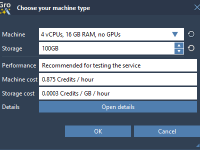
Streamlining Molecular Dynamics with GROMACS Wizard on the Cloud
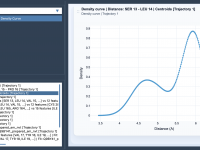
Understanding Radial Distribution Functions (RDF) in Molecular Design.
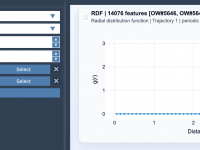
Optimizing Molecular Modeling with Path Attributes in SAMSON
Simplify Your Umbrella Sampling Setup with GROMACS Wizard
Enhance Molecular Modeling with Document Organization in SAMSON
Streamlining Molecular Modeling Tasks with SAMSON’s Interactive Tutorials
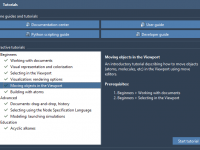
For molecular modelers, learning a new software platform or mastering specific workflows can often feel overwhelming. Whether you’re navigating molecular loading, simulations, or visualizations, sometimes reading static instructions isn’t enough. SAMSON offers a practical solution to this challenge: Interactive Tutorials.…