Streamlining Your Molecular Modeling Workflow with SAMSON Extensions
Simplifying Protein Structure Prediction with AlphaFold-2 in SAMSON.
Accurately predicting protein structures is a critical task for molecular modelers. It provides key insights into biological function, interactions, and potential drug design opportunities. However, protein structure prediction is notoriously complex, often requiring powerful algorithms and significant computational resources. If…
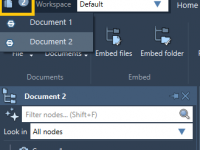
Exploring Folder Attributes in Molecular Modeling: A Guide to NSL
Mastering Camera Views in Molecular Modeling for Effortless Navigation
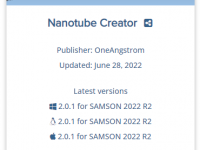
Understanding Compatibility Between SAMSON and Extensions.
Molecular modelers often face a frustrating challenge: ensuring that tools and extensions they rely on are compatible with their current software version. SAMSON, the integrative molecular design platform, addresses this through a well-defined versioning system that brings clarity and predictability…