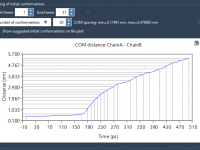
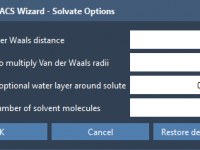
Avoid Solvent Clashes in GROMACS CG Simulations with One Simple Tweak
Filtering Side Chains by Atom Counts with NSL
Exporting DNA Nanostructures for Simulation with Adenita
A Simple Way to Record Molecular Motion Without Coding
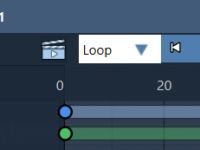
Tracking how molecules move during simulations or interactive presentations can be key when communicating your discoveries. But if you’ve ever tried capturing atom trajectories while modifying structures in real-time—be it docking ligands, assembling molecules, or simulating dynamics—you might know how…
Reclaim Your Interface: Customizing SAMSON the Way You Work
Running GROMACS Simulations in the Cloud Without Configuration Stress
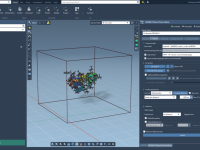
Running molecular dynamics (MD) simulations with GROMACS usually requires not just scientific knowledge, but also technical setup: compiling software, configuring command-line commands, managing dependencies, and securing enough computing resources. This can be a challenge — especially when you just want…