A Quick Way to Reuse Custom GROMACS Parameters in SAMSON
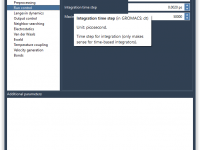
For molecular modelers working with GROMACS, managing molecular dynamics parameters across different simulation steps can be a time sink. If you’re frequently switching between projects, tuning parameters, or experimenting with different equilibration conditions, then manually reconfiguring parameter sets for each…