Writing Molecular Scripts Naturally with SAMSON AI
Narrowing the Docking Search Domain in Hex: When Less Is More
Avoiding Pitfalls When Preparing Coarse-Grained Systems in GROMACS Wizard
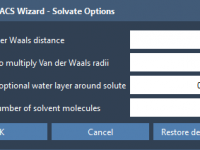
A Quick Way to Define Patterns in Molecular Structures with SMARTS
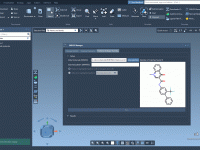
Gradual Reveal: A Practical Tip for Clearer Molecular Presentations
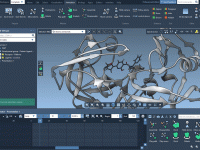
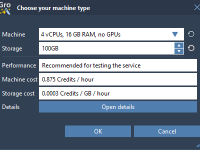
Avoid Costly Surprises: How to Check Your Molecular System Before Launching Cloud Simulations in SAMSON
Making Molecular Visualization More Intuitive with SAMSON’s Visual Models

Molecular modelers often spend significant time adjusting how molecular structures are visualized to interpret and present their data clearly. Whether working with protein secondary structures, electrostatic fields, or electron densities, having intuitive control over visual representation is essential. Yet, many…
Managing Local GROMACS Jobs Without Disrupting Your Workflow
One common challenge for molecular modelers working with GROMACS is dealing with long-running energy minimization calculations. These simulations can temporarily block workflow, especially when computational resources are limited. Fortunately, the SAMSON GROMACS Wizard has integrated local job management capabilities that…