





When modeling structural transitions in proteins, especially for simulations like umbrella sampling or transition path analysis, one recurring challenge for researchers is the correct import and preparation of starting and target conformations. Even minor misalignments, alternate locations, or extraneous molecules can lead to incomplete or interrupted transition paths. In this blog post, we walk through the detailed and often-underappreciated step of importing and preparing your protein conformations in SAMSON before running any interpolation with the As-Rigid-As-Possible (ARAP) Interpolator.
Choosing the Right Structures
For this example, we use the two conformations of Diphtheria Toxin: 1DDT and 1MDT. Each contains chains A and B, but we are only interested in chain A, which represents the portion involved in structural transition.
Fetching Structures in SAMSON
Start by loading the PDB structures directly in SAMSON:
- Open Home > Fetch.
- Enter
1DDT 1MDTin either PDB or PDB (mmCIF). - Click Load.
Once loaded, expand the structures in the Document view. You’ll notice both contain chains A and B, but the interpolation should be performed only on chain A. Including other chains can lead to errors such as non-connected components.
Cleaning and Prepping the Structures
To avoid issues during ARAP path generation, you should strip the structures of unnecessary elements:
- Select
1MDTin the Document view. - Find and delete chain B by clicking the delete icon or pressing Del on your keyboard.
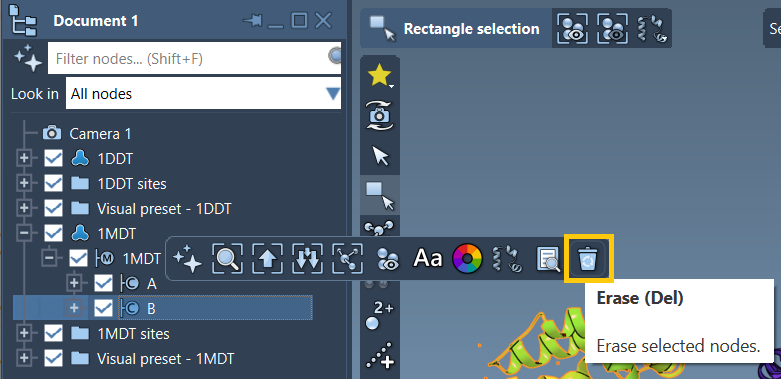
Next, go to Home > Prepare and clean both 1DDT and 1MDT structures. The preparation step removes alternate atom locations, water molecules, ions, and ligands that are often irrelevant for path modeling, but which can cause structural fragmentation.
If you encounter an error like “Cannot proceed because the structure does not make one connected component”, chances are some of those were not removed properly.
Creating Conformations
Now let’s define the conformations that will serve as the start and goal points:
- Select cleaned
1DDT, then go to Edit > Conformation and name it1DDT A. - Repeat the process for
1MDT, naming it1MDT A.
These are critical — ARAP Interpolator models the transition path between these two defined conformations.
Why These Steps Matter
Proper preparation and trimming ensure that only meaningful atoms and bonds are included in your interpolation. It helps:
- Prevent run-time errors in interpolation or simulation extensions.
- Improve biological relevance by focusing analysis on a specific chain.
- Generate smoother transitions thanks to well-aligned, cleaned input.
Skipping structural cleaning or not setting up conformations correctly often results in unusable interpolation paths or incorrect structural alignments, affecting downstream tasks like free energy calculations or animation visualizations.
Conclusion
While it’s tempting to jump straight into interpolation or molecular dynamics simulations, taking the time to prepare and define clean, focused conformations ensures reliable results and avoids frustration later. For a complete walkthrough— including how to run ARAP interpolation and export interpolated trajectories — check the official documentation at this link.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.