





When modeling structural transitions between protein conformations—whether for studying functional motions, setting up umbrella sampling, or preparing molecular dynamics simulations—a common frustration is ensuring that the input structures are properly cleaned, aligned, and ready for downstream tools. One such tool, the As-Rigid-As-Possible (ARAP) Interpolator in SAMSON, allows researchers to generate smooth transition paths between protein structures. But before interpolation can begin, careful structure preparation is essential.
Here is a walkthrough of the initial and often overlooked but essential step: loading and preparing protein conformations for interpolation. We’ll follow an example using 1DDT and 1MDT—two conformations of the diphtheria toxin.
Why preparation matters
Many interpolation methods, including ARAP, assume the input structures are aligned, contain consistent atoms, and exclude unnecessary parts like water molecules, ions, or alternate positions. Errors like disconnected components or mismatched atoms can prevent interpolation or yield meaningless results.
Proper preparation—deleting redundant chains, removing heteroatoms and solvents, and aligning conformers—is what ensures the quality and realism of the computed path. Here’s how to do this in SAMSON.
Step-by-step: loading and cleaning structures
1. Fetch structures in SAMSON:
- Go to Home > Fetch
- Enter
1DDT 1MDTin the PDB field - Click Load
2. Remove unwanted chains: 1MDT contains chains A and B, but we only need chain A for both structures.
- In the Document view, expand
1MDT - Right-click on chain
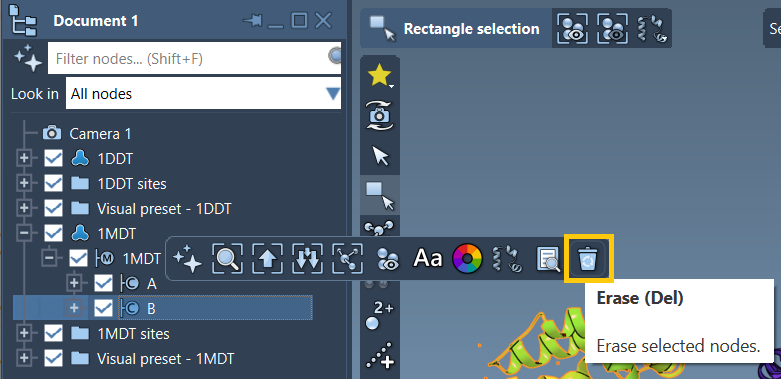
Band delete it - You can use the Del key or click the Erase icon
 when the chain is selected
when the chain is selected
3. Clean the structures: Now that we are only working with chain A in both conformers:
- Navigate to Home > Prepare
- This runs standard cleaning routines: removes water, ligands, ions, and alternate atom locations
- The result: two comparable, clean chains (1DDT A and 1MDT A) ready for conformation creation
Troubleshooting tip: If you get the error “Cannot proceed because the structure does not make one connected component,” run Home > Prepare again—it’s likely due to leftover solvent or ligand atoms.
Once structures are cleaned, you can then save them as conformations in the Edit > Conformation menu (e.g. 1DDT A and 1MDT A), and proceed with interpolation setup.
Conclusion
While it might feel like a small task, correctly setting up your structures at the beginning can save hours of troubleshooting later. By cleaning and simplifying your inputs, tools like ARAP Interpolator can operate reliably, generating realistic transition paths that reflect the biology of the system.
If you’re interested in seeing the full ARAP interpolation workflow, including running the app and exporting results, the full tutorial is available here.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.