





When working on molecular analogs, one of the recurring pain points is figuring out where to introduce chemical modifications. These modifications need to be both chemically justified and relevant for exploring structure-activity relationships. Traditionally, this step requires careful visual inspection or manual scripting, which quickly becomes tedious when you analyze many molecules.
If you’re using SAMSON, the integrative platform for molecular design, you can now handle this more efficiently using the SMILES Manager extension. Let’s zoom into a specific feature: the ability to search for atomic patterns in your molecule using SMARTS, an expressive language used by cheminformaticians to specify substructures.
In this walkthrough, we’ll look at how to rapidly identify and select a recurring pattern in your molecules—for example, aromatic hydrogens—using the [cH] SMARTS pattern. This allows you to define positions suitable for chemical modifications, such as creating positional analogs.
Why this matters
Before performing any substitution, you need to be precise about where in the molecular structure the changes will occur. Pattern recognition with SMARTS ensures positional accuracy. It’s a great way to automate what would otherwise be a labor-intensive task, especially when dealing with larger molecule libraries or when planning systematic modifications across several sites.
How to search with SMARTS in SAMSON
First, load your molecule in SAMSON. You can paste its SMILES code or select it from your existing document. Once done, move to the SMILES Manager interface and locate the field where you can enter a SMARTS query.
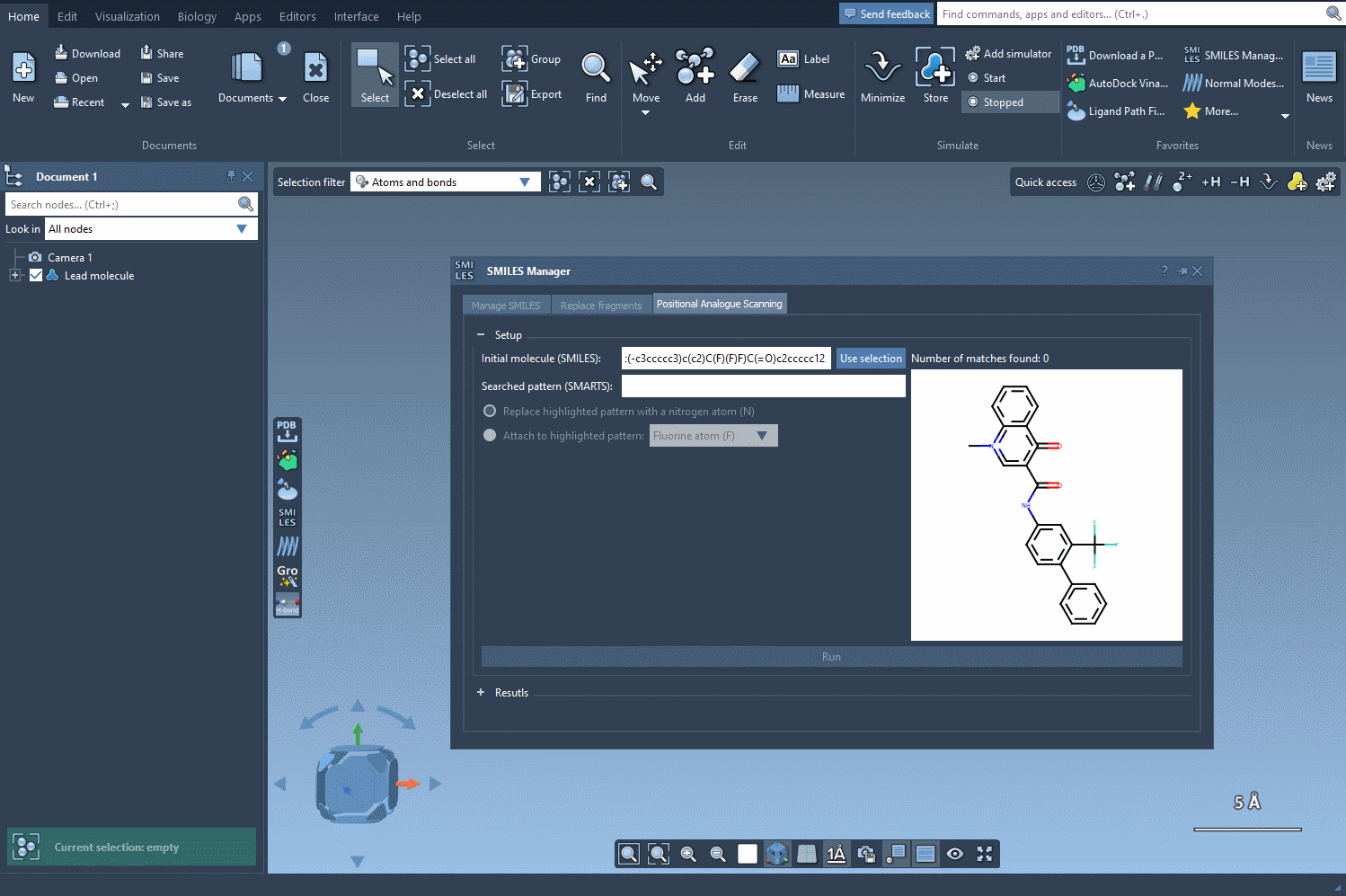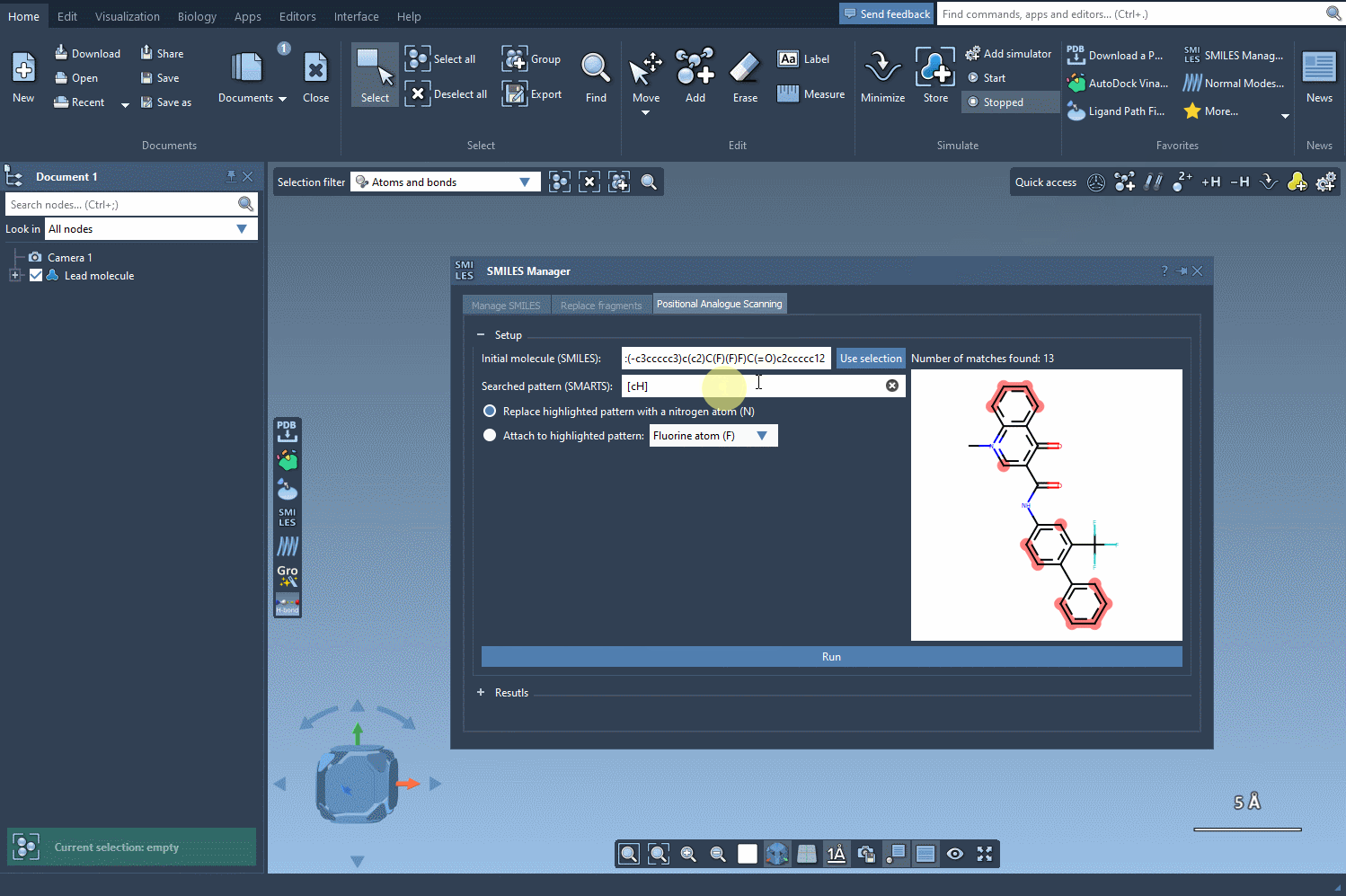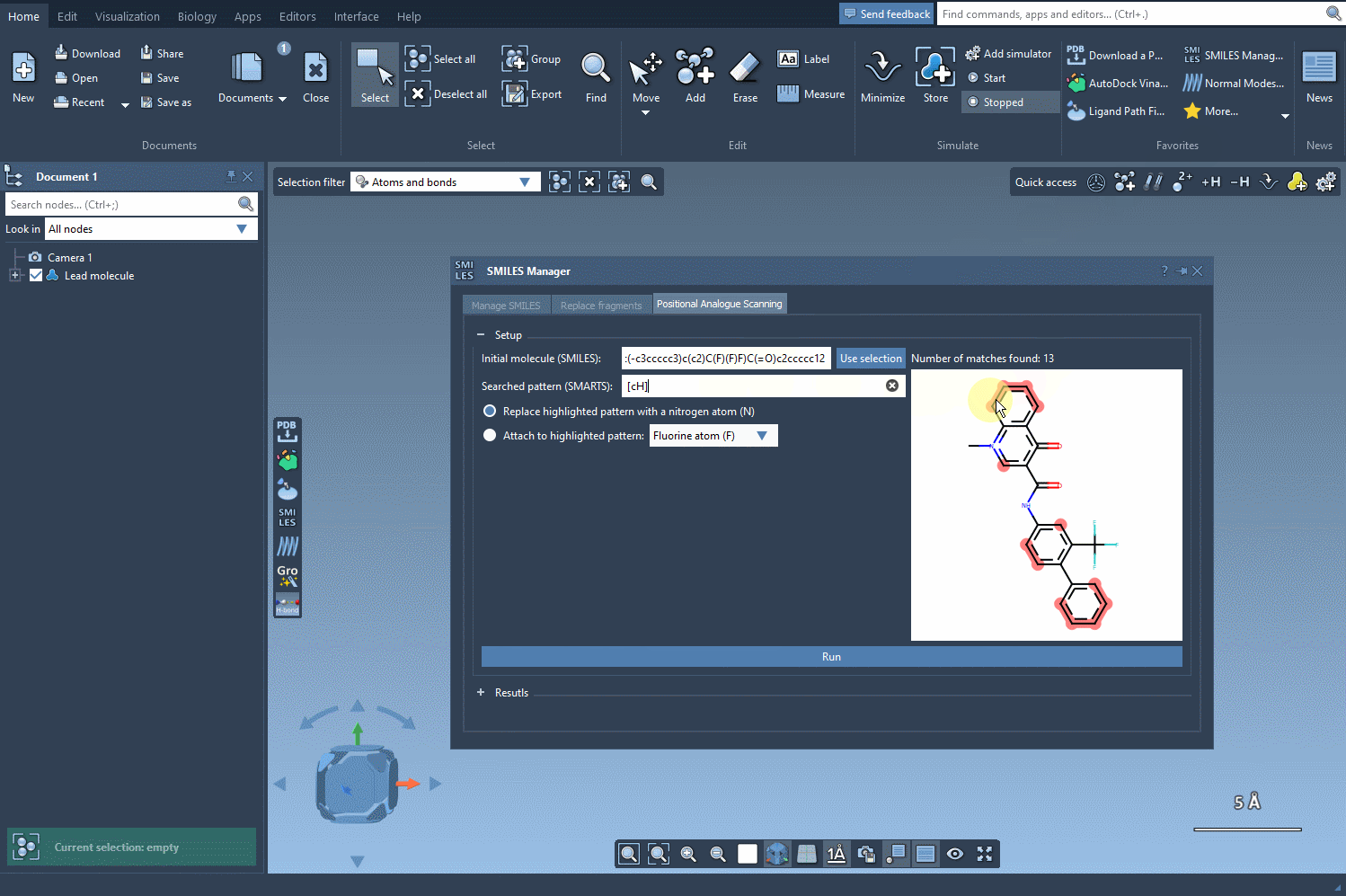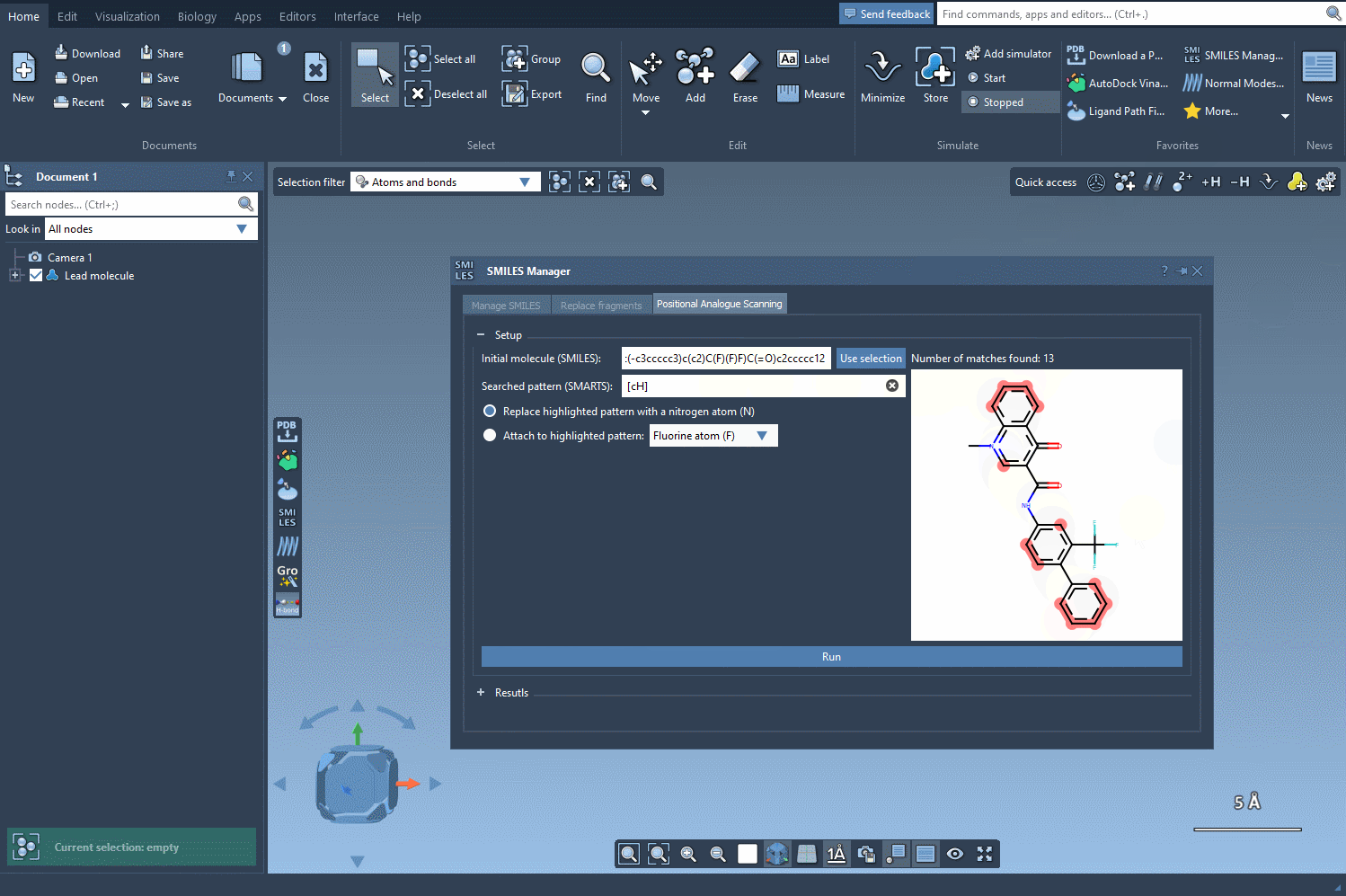
Type in [cH]—which stands for an aromatic ring carbon with one hydrogen attached. This is a common pattern in drug-like molecules and a good starting point for testing substitutions.
SAMSON will highlight all atoms in the structure matching this pattern. As the animated gif below shows, it’s an instant process and visually intuitive:
What can you do next?
Once the relevant pattern positions are detected, you have the flexibility to:
- Replace each instance with other atoms like nitrogen (N) or groups like CH3
- Attach new groups instead of replacing the atom
- Generate all possible analogs from these substitutions for further analysis
This pattern-based atom selection becomes the foundation for generating a diverse analog series, which you can later evaluate for binding affinity, docking, or interaction with a target protein—directly in SAMSON.
When should you use this?
If you’re running a structure-activity relationship (SAR) study or preparing molecules for high-throughput docking experiments, using SMARTS patterns offers greater control. It ensures that your modifications occur at chemically meaningful positions—and that’s essential when trying to understand activity trends across analogs.
To learn more about how to use SMARTS search and positional analogue scanning in SAMSON, check out the official documentation page: Perform Positional Analogue Scanning using the SMILES Manager.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download it at https://www.samson-connect.net.