





When working with small molecule design, one of the most common and tedious challenges is identifying chemical substructures you want to modify, replace, or expand upon. Whether you’re trying to explore structure-activity relationships (SAR) or optimize molecular properties, pinpointing the right atoms or groups to tweak can be time-consuming—especially when doing this manually or across many analogs.
In SAMSON, this process has been streamlined thanks to the SMILES Manager extension. One of its helpful features is the ability to automatically detect substructures in a molecule using SMARTS patterns. Let’s take a closer look at how this works and how you can benefit from it.
Imagine you have a drug-like molecule and you’re interested in exploring the effect of replacing certain aromatic hydrogens with small substituents. Instead of manually inspecting the molecule and interpreting its connectivity, you can simply input a SMARTS pattern into the interface and receive immediate visual feedback on which atoms match your pattern.
Step-by-step: Highlighting SMARTS Patterns
In the SMILES Manager interface, once you’ve selected your molecule of interest (either by pasting the SMILES code or selecting it in the document and clicking Use selection), you’ll find a field where you can input your desired SMARTS pattern.
For aromatic hydrogens, the SMARTS pattern [cH] is used. When this is entered, all matching atoms in the molecule are automatically highlighted:
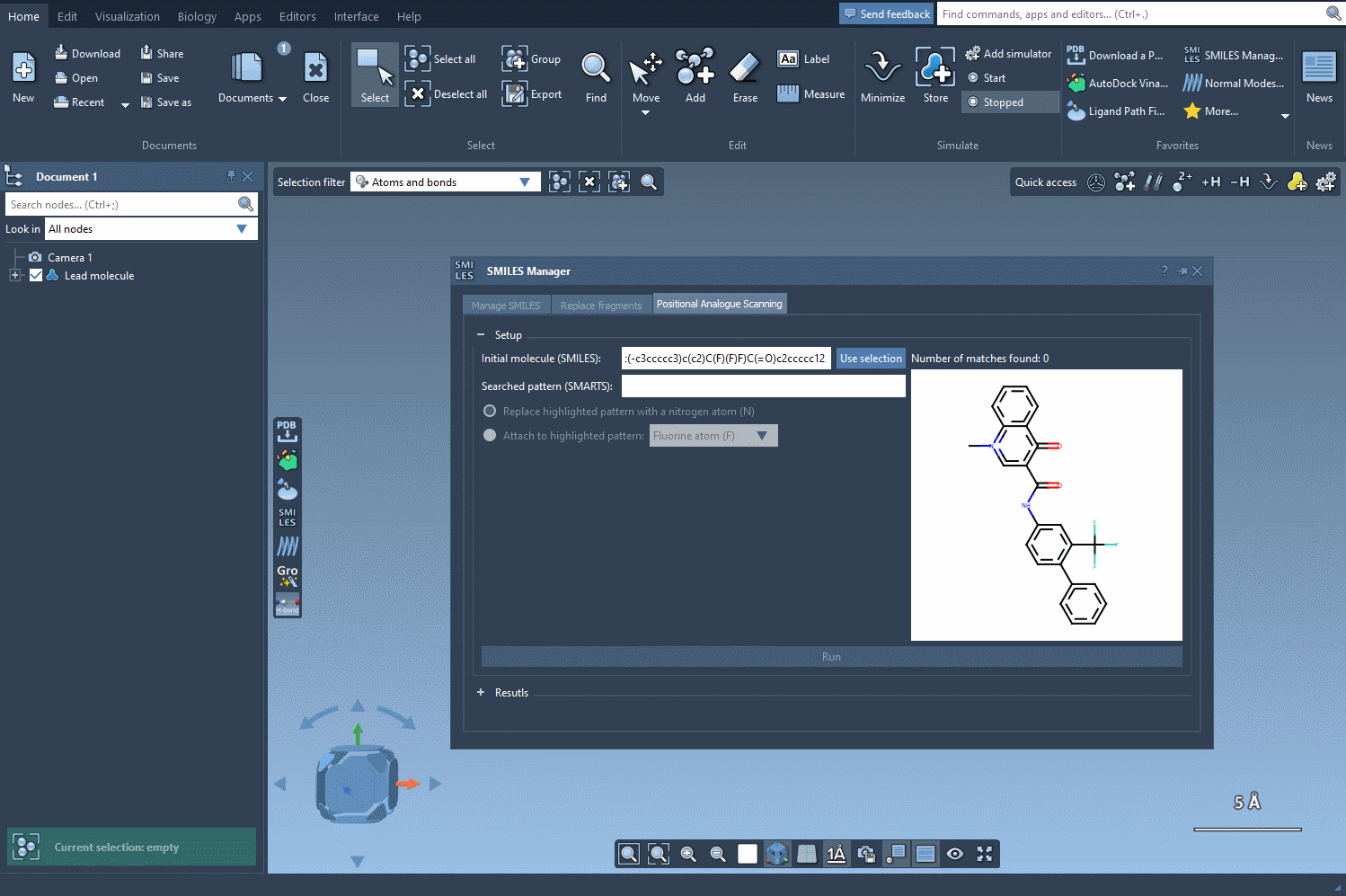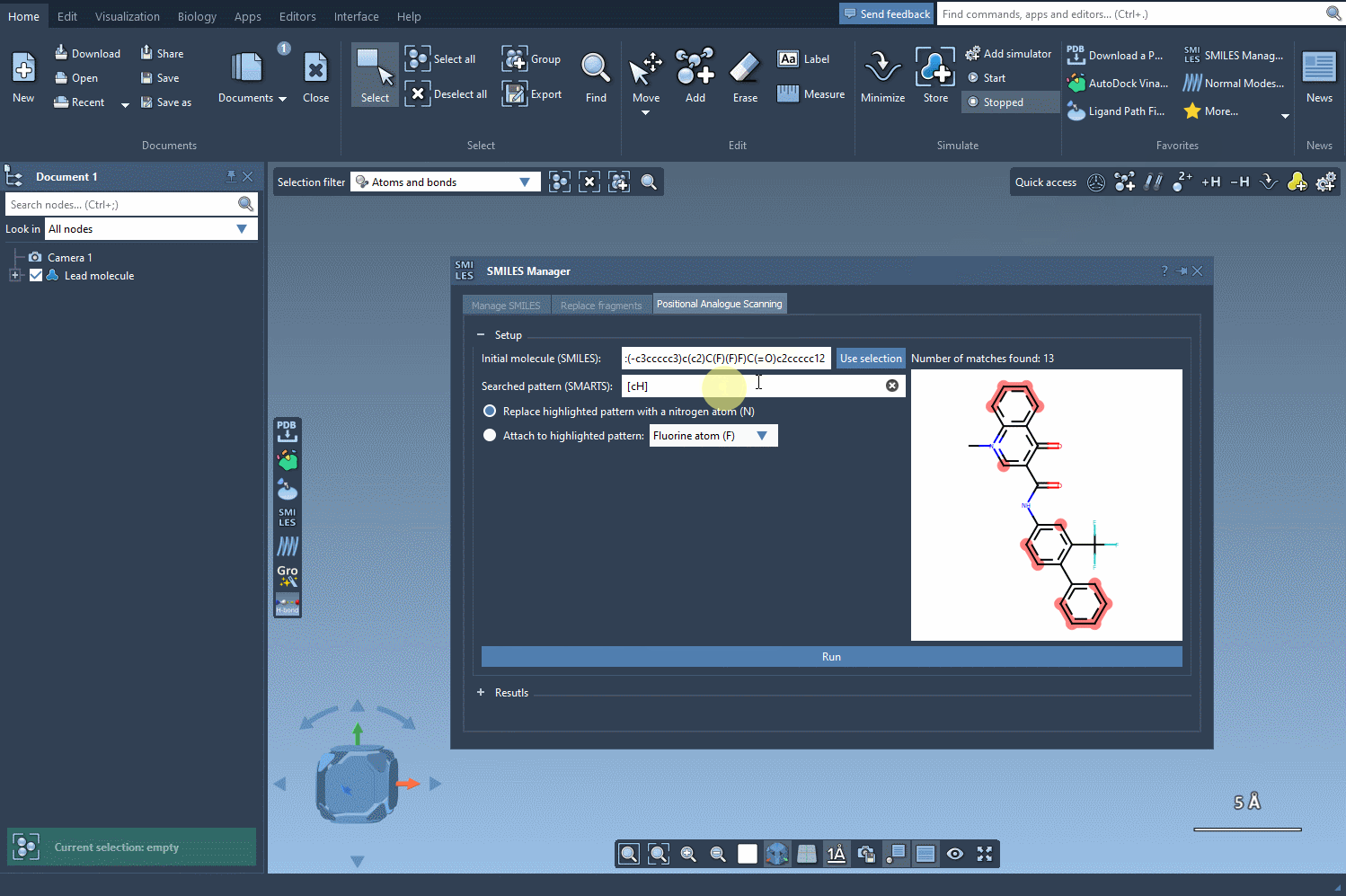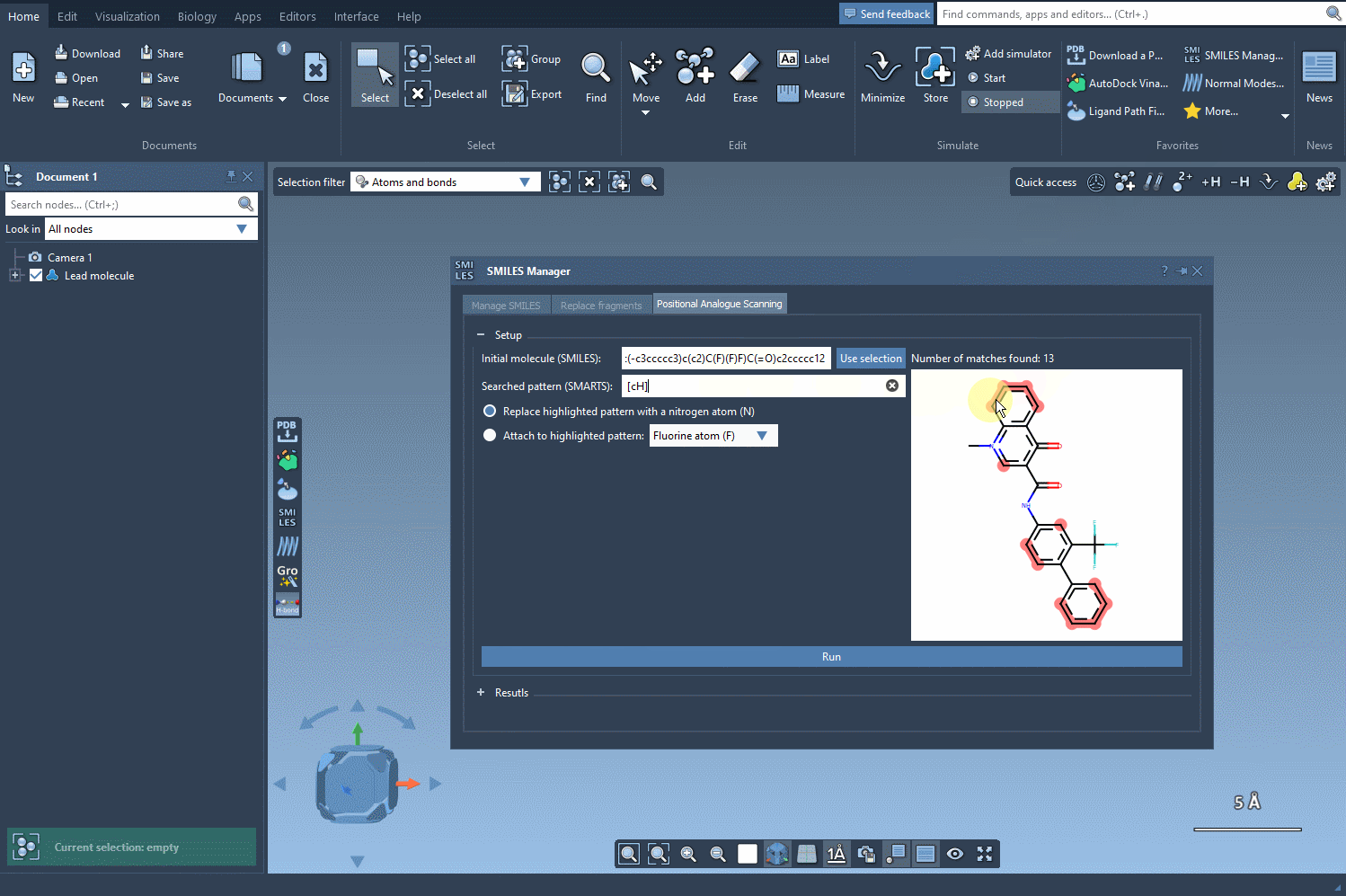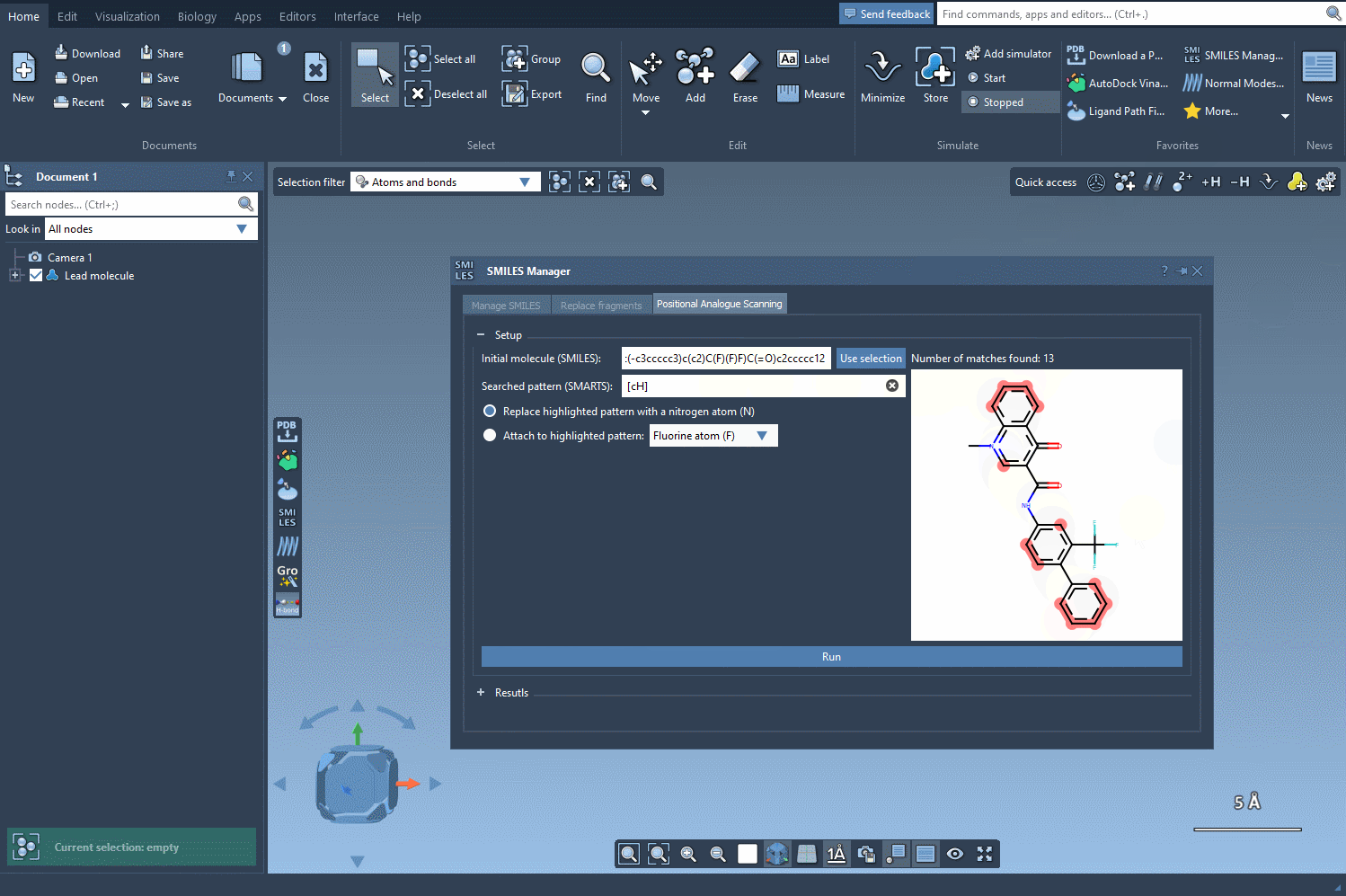
This visualization gives you immediate insight into where your substructures of interest occur. The matches are clearly marked, making it much easier to decide which locations might be most amenable to structural modifications.
Why Is This Useful?
- Saves time: No need to inspect each bond and atom manually.
- Reduces errors: Get consistent matches across molecules using formal definitions.
- Flexible pattern search: Use simple or complex SMARTS to isolate specific chemical groups or environments.
Once you’ve identified the relevant atoms, you can proceed to replace them with alternative groups (e.g., N, F, CH3), thus generating positional analogs quickly. This forms the basis for Positional Analogue Scanning, a method that helps assess the functional impact of small changes on molecular properties like binding affinity or lipophilicity.
Benefits for Molecular Modelers
If your work involves lead optimization or compound library design, being able to identify and act on specific structural motifs within seconds can dramatically accelerate your workflow. This SMARTS-enabled substructure matching isn’t limited to aromatics—you can define any chemical feature you like, from hydrogen bond donors to sp3 carbon atoms.
To sum up, the SMARTS-based pattern discovery in SAMSON’s SMILES Manager can boost your productivity by helping you focus immediately on modifiable sites in your molecules. It’s a simple yet valuable tool that eliminates guesswork and supports rational compound design.
To learn more—including how to run full analogue scans and study their 3D structures—read the full tutorial: Perform Positional Analogue Scanning using the SMILES Manager.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.