





Molecular modelers often struggle with capturing realistic transitions between protein conformations, especially for complex systems. Such transitions are essential for tasks like conformational analysis, reaction coordinate generation, or setting up free energy simulations. If you find this process time-intensive and challenging, the ARAP Interpolator from SAMSON offers an efficient solution. Here’s how you can use it to generate smooth, biologically meaningful transition paths in just a few steps!
The Pain Point: Complex Conformational Transitions
Modeling structural transitions between protein states often involves manual alignment or computationally heavy techniques. This process can be cumbersome and prone to errors, especially when aligning intricate systems or interpreting intermediate steps. Researchers need a faster, streamlined method that maintains biological relevance.
The Solution: ARAP Interpolation in SAMSON
The ARAP Interpolator in SAMSON simplifies the creation of protein conformation pathways. It generates a smooth, biologically meaningful transition between two protein conformations in seconds, using the principle of As-Rigid-As-Possible interpolation. This process minimizes deformation while maintaining structural integrity.
Key Benefits of ARAP Interpolation
- Generate smooth, continuous structural transitions rapidly.
- Visualize and export intermediate conformations for further analysis.
- Align and organize conformers using a biologically meaningful geometric model.
- Build realistic reaction coordinates for free energy simulations.
A Step-By-Step Guide to Generate Transition Paths
Before You Start
- Ensure you are logged into SAMSON Connect.
- Install the ARAP Interpolator from its extension page.
- Restart SAMSON to activate the extension.
Step 1: Prepare the Protein Structures
Fetch your target structures in SAMSON by navigating to Home > Fetch. For instance, fetch 1DDT and 1MDT from the PDB database. These represent two conformations of the Diphtheria Toxin. Our focus is on chain A:
- Expand the
1MDTmodel in the document view. - Delete chain
Busing the Del key or the “Erase” toolbar option.
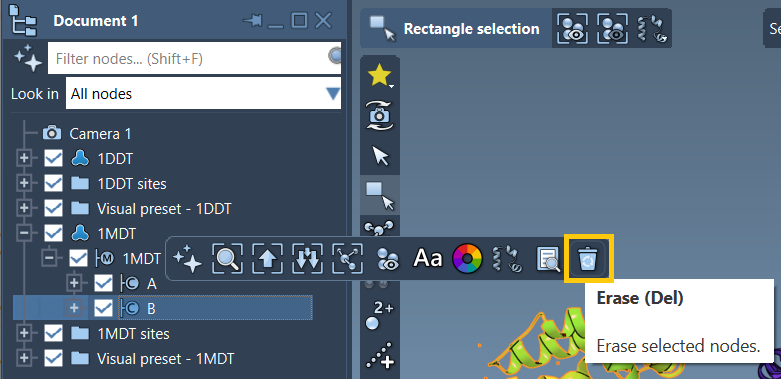
Finally, clean your structures from any unnecessary components (e.g., water, ligands) by running Home > Prepare.
Step 2: Define Conformations
The ARAP Interpolator works by transitioning between conformations:
- In the document view, select
1DDT, click Edit > Conformation, and name it1DDT A. - Repeat this for
1MDT, naming its conformation1MDT A.
These designated conformations serve as the start and goal of your transition pathway.
Step 3: Configure and Run ARAP Interpolation
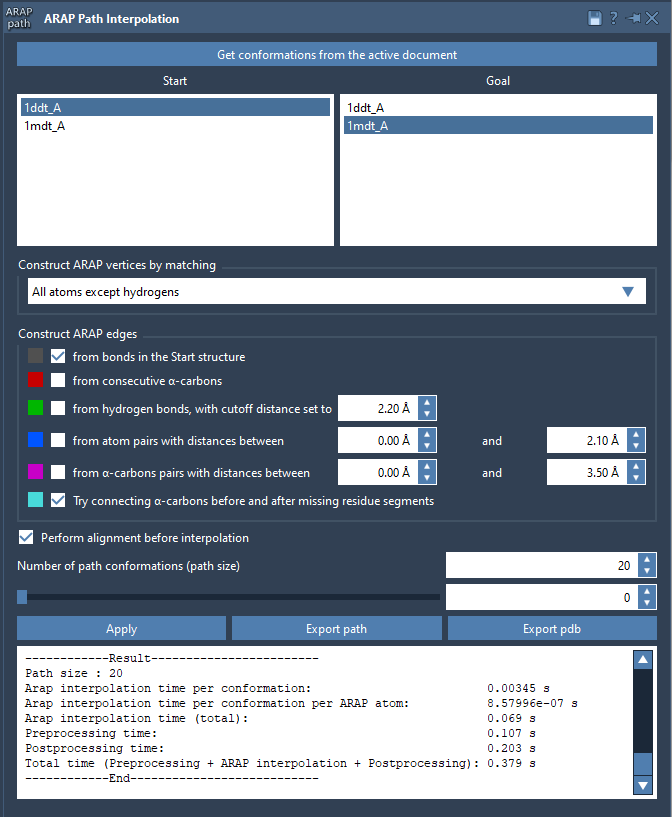
Open the Interpolation App via Home > Apps > Biology > ARAP Path Interpolation and configure the following steps:
- Choose Conformations: Select your start (
1DDT A) and goal (1MDT A). - Matching Atoms: Choose “All except hydrogens” to align non-hydrogen atoms.
- Construct Edges: Check “from bonds in the Start structure” and “Try connecting α-carbons before and after missing residue segments.” This ensures proper path continuity.
- Enable Perform alignment before interpolation.
- Set the Number of path conformations to 20 to divide the transition into 20 frames.
Once everything is set, click “Run” to compute the interpolation. Visualize the results using the slider in the app interface.
Step 4: Analyze and Export Results
After computation, use the app to analyze the path or export it for further use:
- Export paths into trajectory objects or PDB files.
- Use visual aids like edge construction models to understand pathway logic.
Transition paths generated via ARAP Interpolation are ideal for downstream applications like molecular dynamics or umbrella sampling.
Learn More
To delve deeper into using ARAP Interpolation for protein transitions, visit the complete documentation at this link.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can access SAMSON at SAMSON Connect.