





When building or refining protein structures—whether from experimental data, homology modeling, or simulations—one common challenge is spotting conformational outliers that may introduce artifacts in downstream analyses. A residue that strays too far from allowed dihedral angles can destabilize simulations, distort predictions, or mislead active site analyses. If you’ve ever spent hours debugging structural oddities, you know how helpful it would be to catch these issues early on.
The Interactive Ramachandran Plot Extension in SAMSON provides a straightforward visual tool to evaluate backbone geometry—and more importantly, interactively correct problematic residues with either a drag gesture or a structure-aware editor. Here’s how you can integrate this tool into your refinement workflow to quickly identify and fix strained regions in your protein models.
Step 1: Load and Visualize Your Structure
Open a protein structure in SAMSON. You can use a known structure, such as PDB ID 1YRF:
- Go to Home > Fetch
- Type
1YRFand click Load
Step 2: Access the Ramachandran Plot
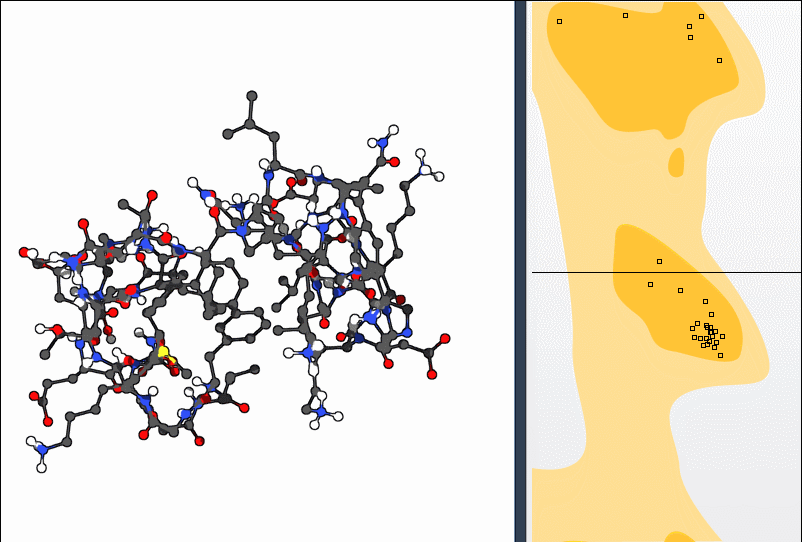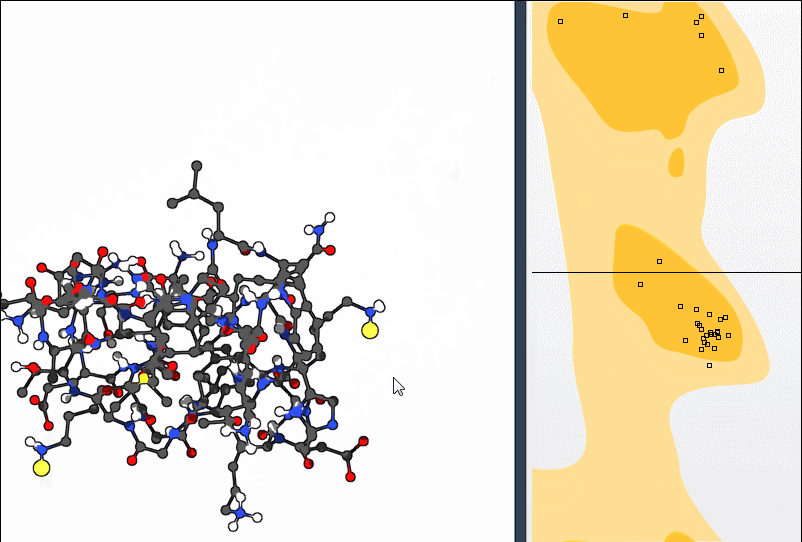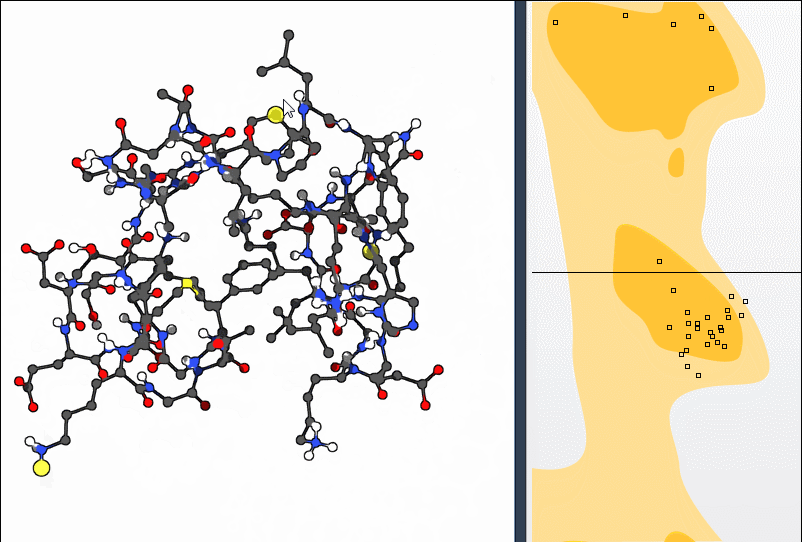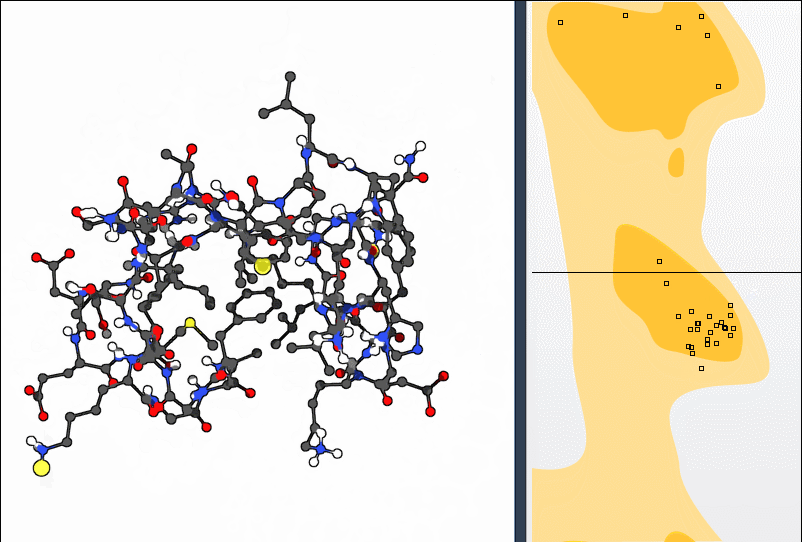
Navigate to Home > Apps > Biology > Ramachandran plot to open the extension. Click the Update button to generate the full φ-ψ backbone angle plot for your structure:
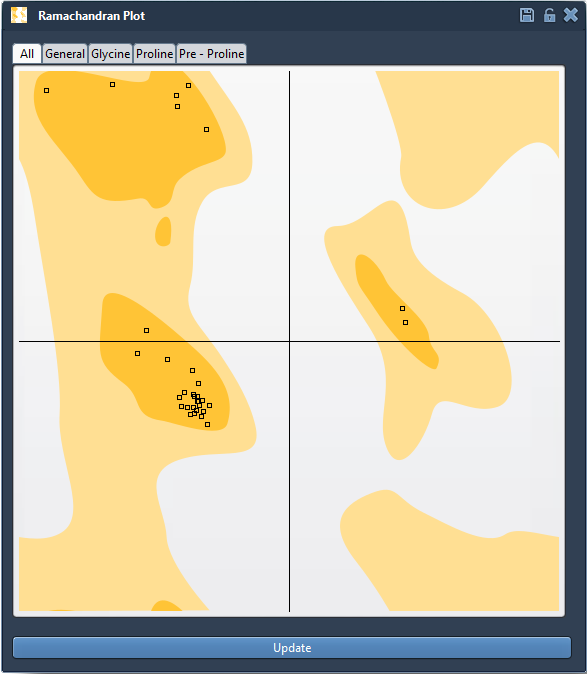
- Yellow areas indicate energetically favorable conformations
- White areas show disallowed backbone geometries
If you see any dots in the white regions, you’ve likely identified an outlier that could be worth investigating.
Step 3: Inspect and Interact
Click on a specific residue dot in the plot. You’ll immediately see it highlighted in the 3D viewport. The φ and ψ values for the residue also appear in the status bar:


Step 4: Fix Outliers (Two Methods)
Option 1: Drag and Drop
- Simply drag the dot in the plot to shift the φ and ψ values
- The 3D protein structure updates in real-time
- Undo with Ctrl/Cmd + Z if needed
Option 2: Use the Twister Editor
- Select the Twister Editor from the left-hand toolbar:
- Manually rotate the backbone in the 3D view and watch the plot update live
Both approaches allow you to bring a residue back into a favorable dihedral region without manually entering angles or switching between separate tools.
Why This Matters
Residue outliers are often overlooked until simulations become unstable, or predictions diverge from expected biology. The ability to quickly spot and correct these outliers interactively saves time and prevents downstream errors. You can even apply this as a cleanup step before exporting your model or submitting it for further study.
For more guidance, including how to combine this approach with normal mode analysis for broader structural refinement, see the full documentation: https://documentation.samson-connect.net/tutorials/ramachandran/ramachandran-plot/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.