





For molecular modelers, one common challenge is setting up simulations efficiently without getting bogged down by complicated variables. Enter the Universal Force Field (UFF), an interaction model in SAMSON designed to streamline the process of simulating molecular models. Whether you’re researching new compounds, testing structures, or teaching molecular dynamics, UFF provides a straightforward yet robust way to kickstart molecular simulations. In this post, we’ll focus on the essential steps to set up UFF in SAMSON and get your first simulation running quickly and efficiently.
What is the Universal Force Field?
The Universal Force Field, proposed by Rappe et al., covers elements across the periodic table, making it widely applicable to diverse molecular systems. SAMSON’s implementation of UFF simplifies simulation setup by automatically perceiving molecular structure to compute bonds, bond orders, and atom types. This automation lets you immediately apply UFF to simulate or optimize your molecular system.
Steps to Set Up UFF
Here’s a concise guide to getting started with UFF in SAMSON:
- Prepare your molecular system: First, open the document containing the molecular system you want to simulate using UFF. If you’re a beginner, consider starting with a simple system and default settings for clarity.
- Add the UFF simulator: Add a UFF simulator by navigating to
Edit > Simulate > Add simulator. Alternatively, use the shortcutCtrl + Shift + M(Windows) /Cmd + Shift + M(Mac). - Select UFF: Choose “Universal Force Field” from the list of interaction models that appears.
- Choose a state updater: Specify the state updater for your simulation, such as the Fast Inertial Relaxation Engine (FIRE), to optimize geometry or relax the system.
- Click OK when you’re done to proceed with UFF setup.
The UFF Setup Window
Once the UFF setup window appears, you’ll have a few key options to configure:
- Use existing bonds: Enable this feature if you already have defined bonds you want to retain. Otherwise, UFF will compute bonds automatically based on atom types and positions.
- Initialize the system: After making your selection, click OK. UFF then performs the following operations automatically:
- Computes covalent bonds if “Use existing bonds” is unchecked.
- Determines bond orders and assigns atom types.
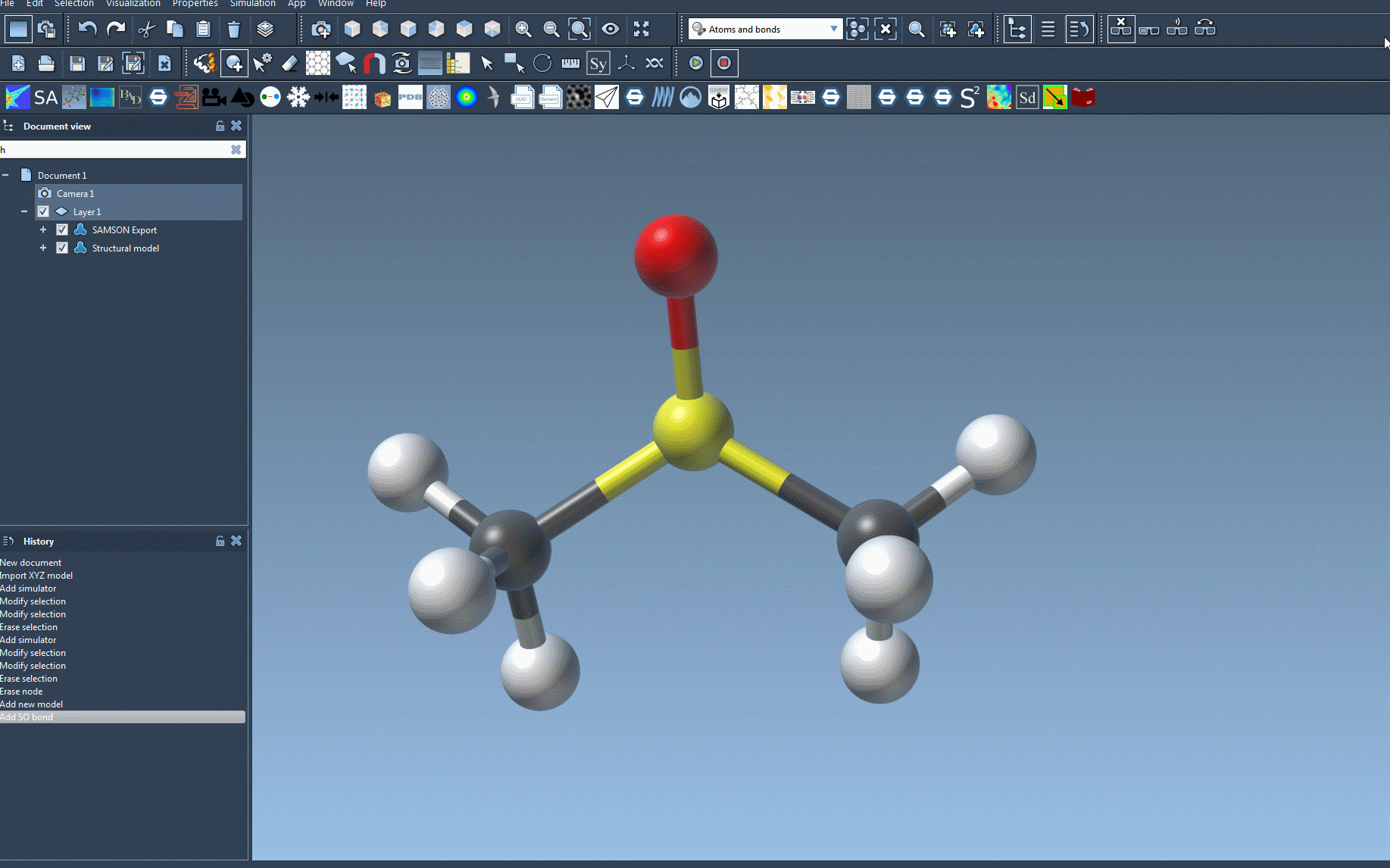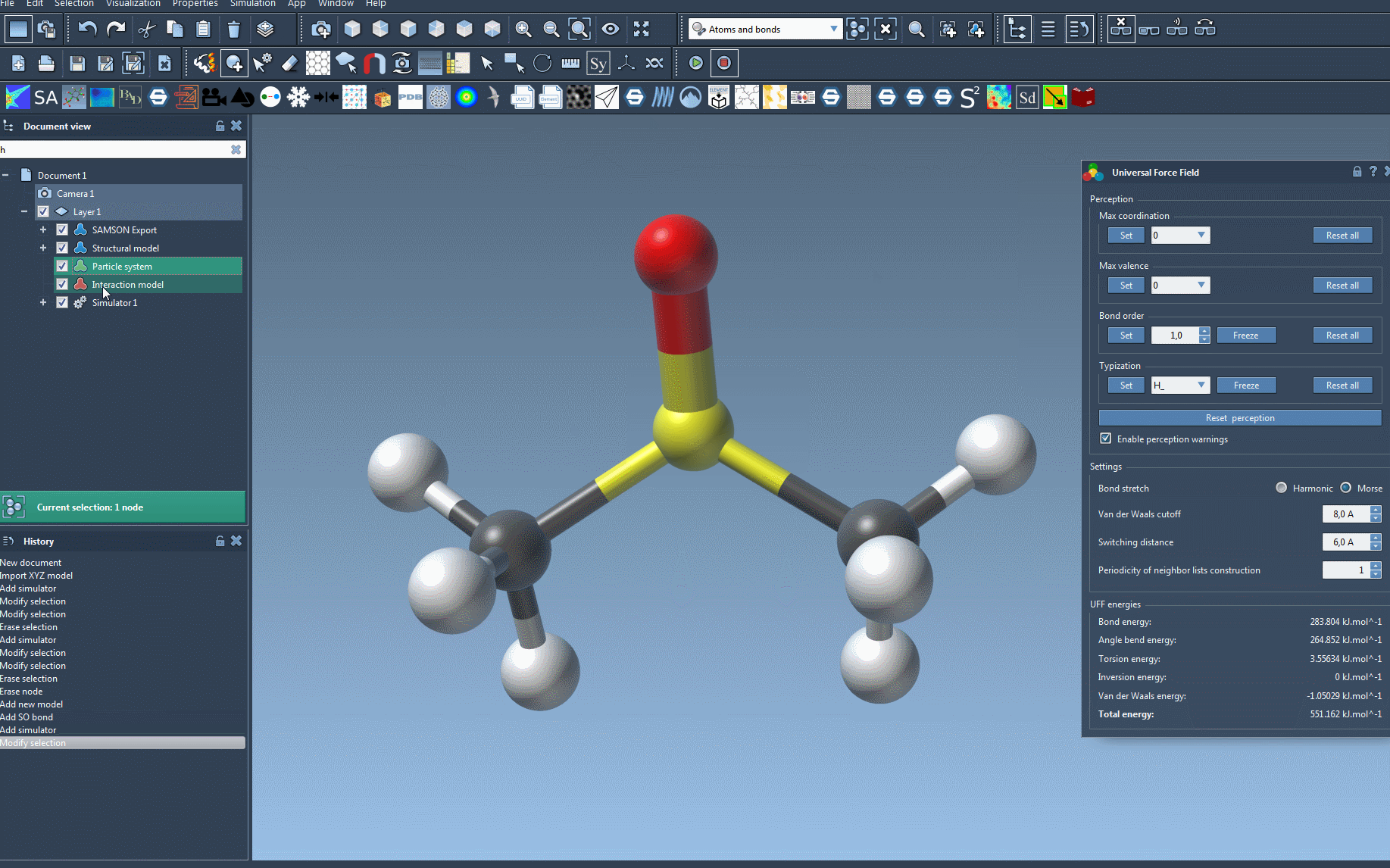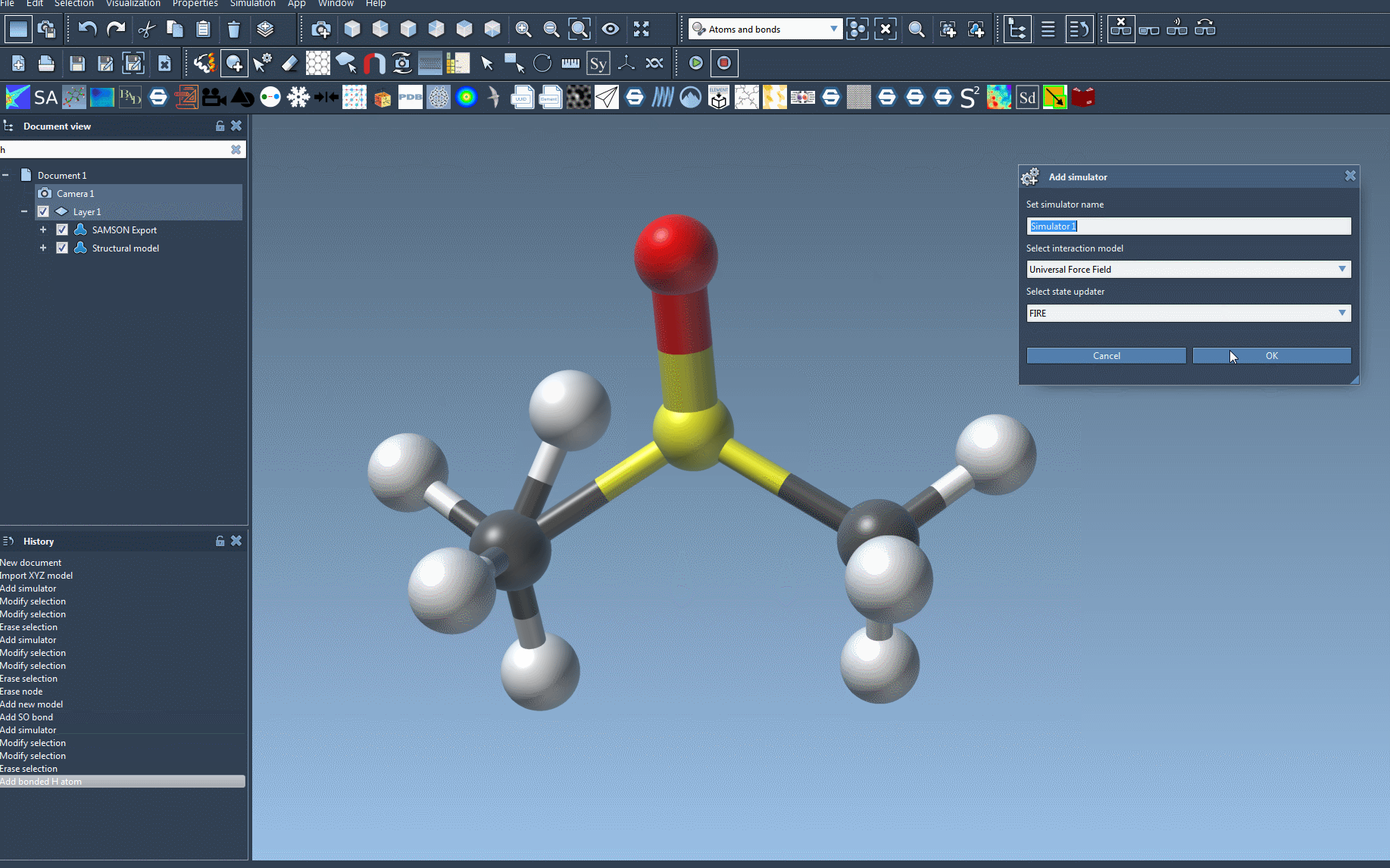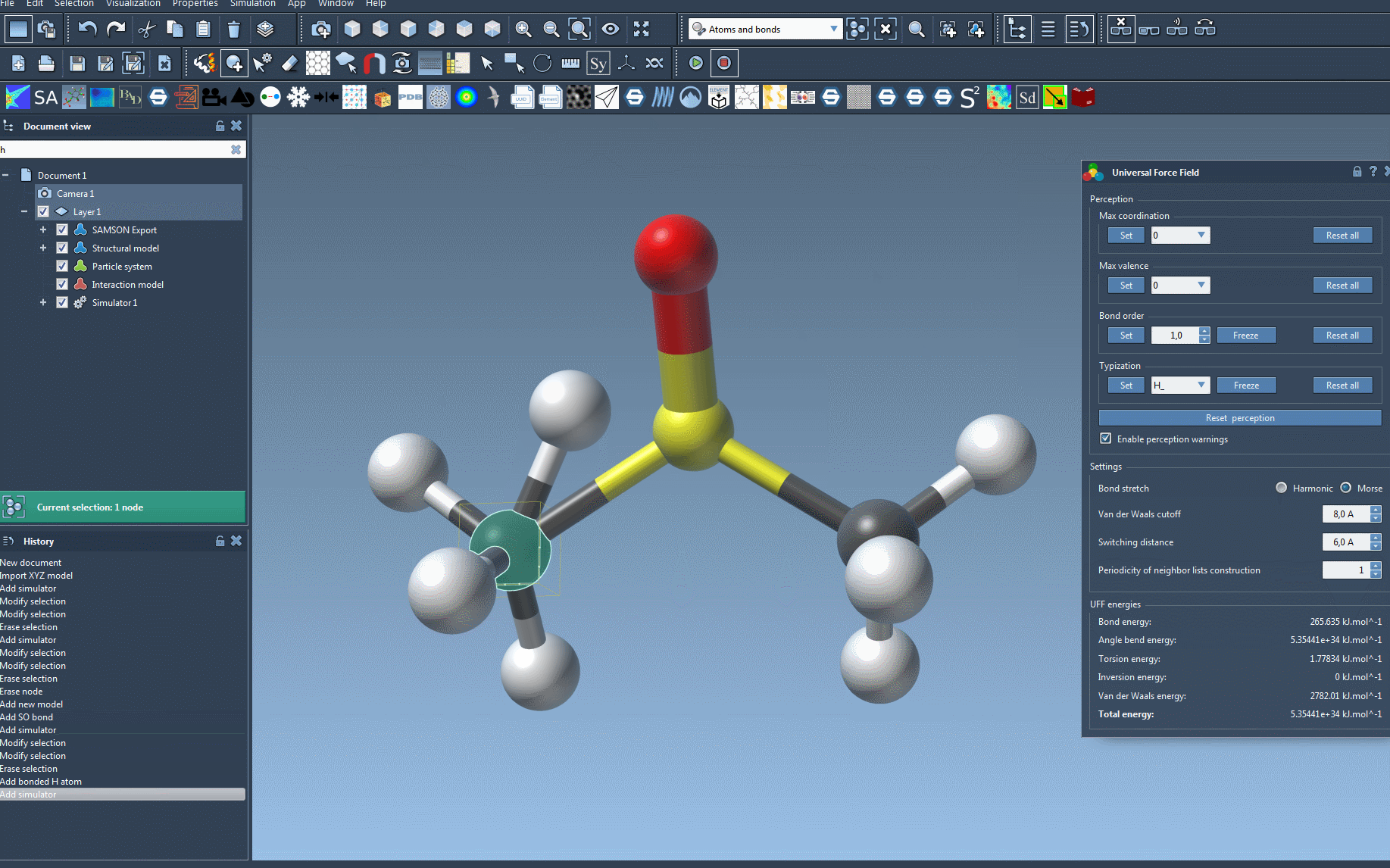
If there are any inconsistencies, SAMSON will notify you with a warning message, so you can review and address potential issues before proceeding.
Running Your First UFF Simulation
Once the initialization is complete, you are ready to simulate:
- Click
Edit > Simulate > Startto begin the simulation.
Your molecular system will now evolve based on the Universal Force Field. The UFF interface will display energy for each term, as well as the total energy in real-time. You can even interactively move atoms and observe how UFF adapts the positions of other atoms in response, demonstrating the physical interactions at play.
A Great Tool for Molecular Modelers
The UFF module provides a seamless way to set up and test molecular systems, even with minimal prior experience. With its automated atom type and bond computation, you can focus less on tedious setup details and more on exploring your molecular model’s behavior. For those encountering difficulties, SAMSON offers various resources like its forum and interactive tutorials to support your simulation journey.
For detailed instructions and additional features like parameter customization, please refer to the official documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Learn more and get SAMSON at SAMSON Connect.