





For molecular modelers diving into simulation or molecular design work, setting up an efficient force field is crucial. However, it can also feel intimidating or overly detailed. If you’ve been struggling with applying force fields effectively, SAMSON offers a straightforward way to integrate the Universal Force Field (UFF) into your workflow. Here’s a step-by-step approach to making it work smoothly.
Why Choose UFF?
The Universal Force Field, introduced by Rappe et al., provides wide applicability because it covers the entire periodic table, from organics to heavy metals. This makes it perfect for versatile modeling scenarios. Setting it up in SAMSON also benefits from its unique automatic perception scheme, which calculates bonds, bond orders, and atom types for your molecular system seamlessly.
How to Set Up UFF in SAMSON
Setting up UFF in SAMSON is straightforward. Here’s how you can get started:
- Open a molecular document: First, choose the molecular system you’d like to analyze or simulate.
- Add a simulator: Navigate to
Edit > Simulate > Add simulator. Alternatively, you can use the keyboard shortcutCtrl + Shift + M(orCmd + Shift + Mon Mac). - Select UFF: From the list of interaction models, click on Universal Force Field.
- Choose a state updater: Select the state updater you prefer for simulation tasks, such as the Fast Inertial Relaxation Engine (FIRE), which ensures smooth energy minimization.
- Confirm settings: Press the OK button to proceed.
At this point, the UFF setup window will appear, providing additional options:
- Use existing bonds: If your molecular system already has defined bonds, you can check this option. Otherwise, UFF recalculates everything based on atom positions and types.
- Automatic Initialization: After clicking OK, UFF computes covalent bonds, bond orders, and atom types to initialize the system.
If any structural inconsistencies are detected, SAMSON will alert you with a detailed warning screen so adjustments can be made promptly.
Run and Explore Simulations in Action
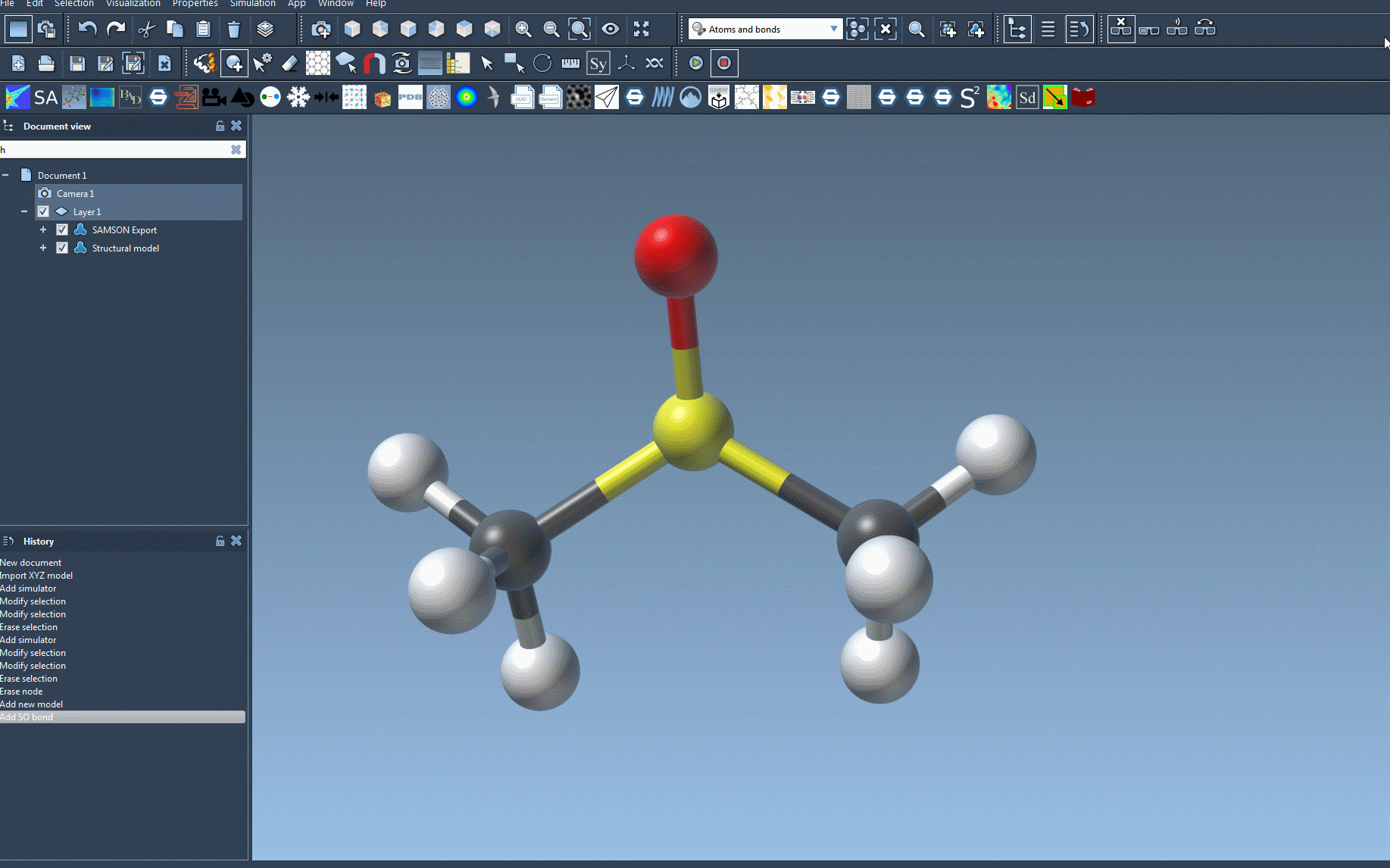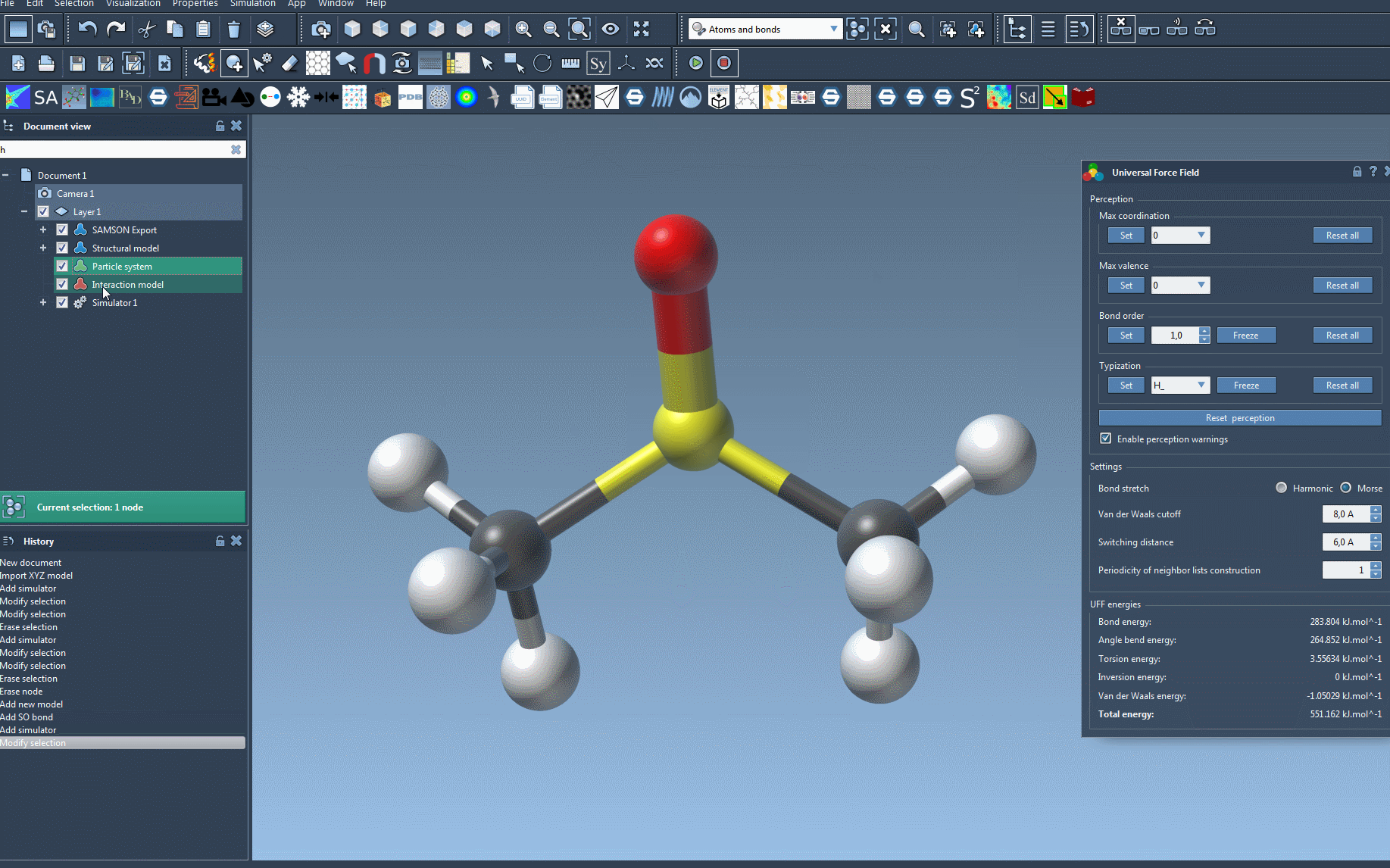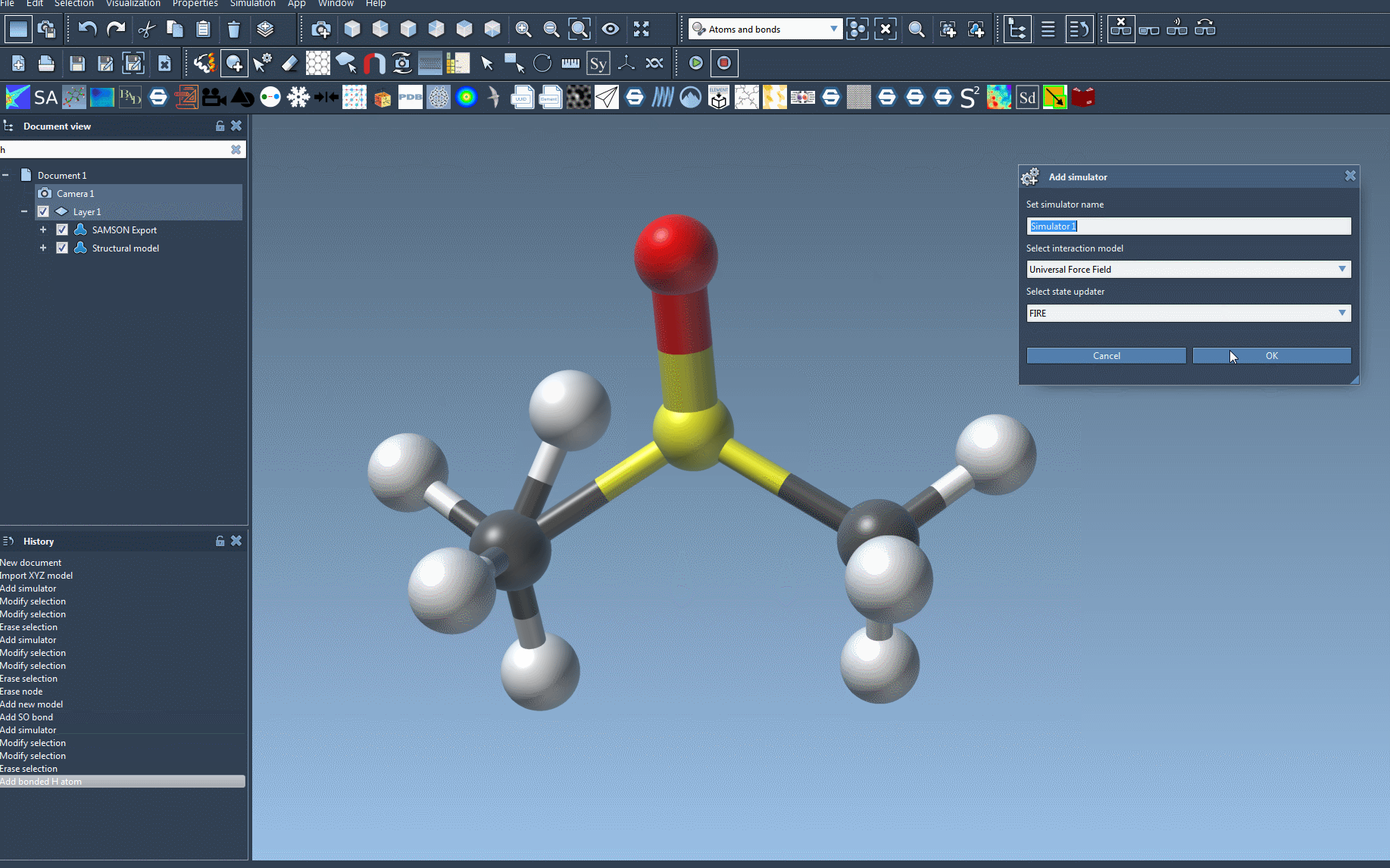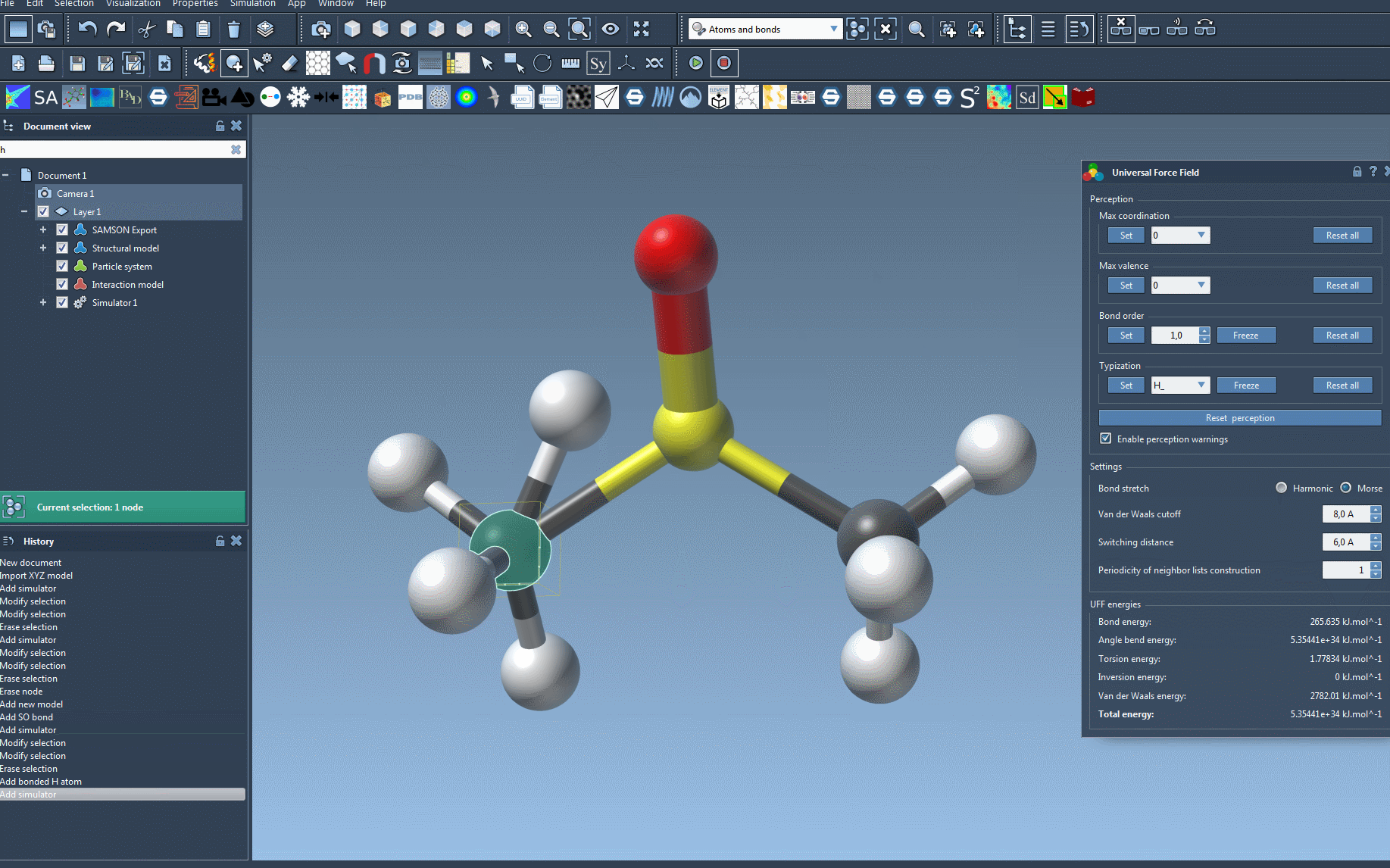
Once your setup is complete, running the simulation is as simple as selecting Edit > Simulate > Start. During the simulation, you’ll be able to monitor the energy terms and total energy in the UFF interface. It also allows you to move atoms interactively and observe how the force field adjusts atom positions dynamically as a response. This capability is particularly engaging and educational, helping creators understand molecular behavior in real time.
Customization options let users adjust parameters like bond stretch interactions (switching between Harmonic and Morse), van der Waals cutoff distance, and even periodicity for neighbor lists during simulation, producing specific, tailored results.
Conclusion
With SAMSON’s UFF implementation, you don’t need to spend extra hours manually tuning forces or guessing atom types. By automating much of the molecular perception process while still leaving room for customizations, SAMSON ensures flexibility without sacrificing efficiency. To learn more, explore the official UFF tutorial here.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.