





One of the biggest challenges for molecular modelers is efficiently simulating and analyzing molecular systems with accurate force field models. The Universal Force Field (UFF) is a widely used interaction model that offers a robust solution for simulating molecular mechanics and dynamics across the periodic table. In this post, let’s explore how you can effortlessly run UFF simulations using the SAMSON integrative molecular design platform, a feature-packed tool designed for researchers in nanoscience, chemistry, and biology.
What is the Universal Force Field (UFF)?
UFF is a parameterized force field developed for molecular mechanics simulations across the periodic table. It computes essential interactions such as bonds, bond orders, and atom types, allowing modelers to accurately represent molecular systems. SAMSON’s UFF implementation is particularly convenient as it includes an automatic perception scheme that deduces the molecular structure, making the setup process highly intuitive.
How to Get Started with UFF Simulations
Running a simulation in SAMSON using UFF is straightforward. Here’s how to set it up:
- Open the molecular system you want to simulate in SAMSON.
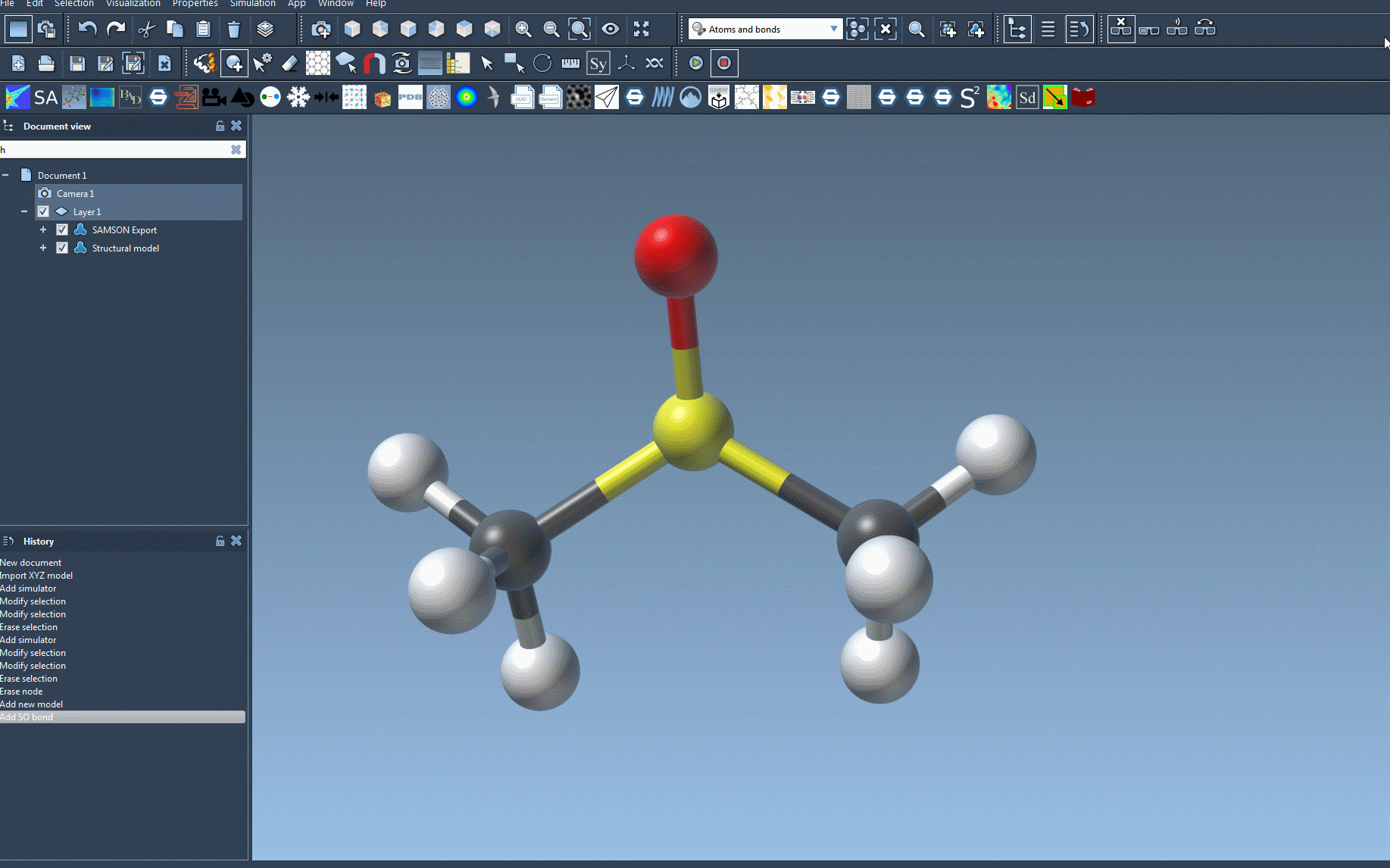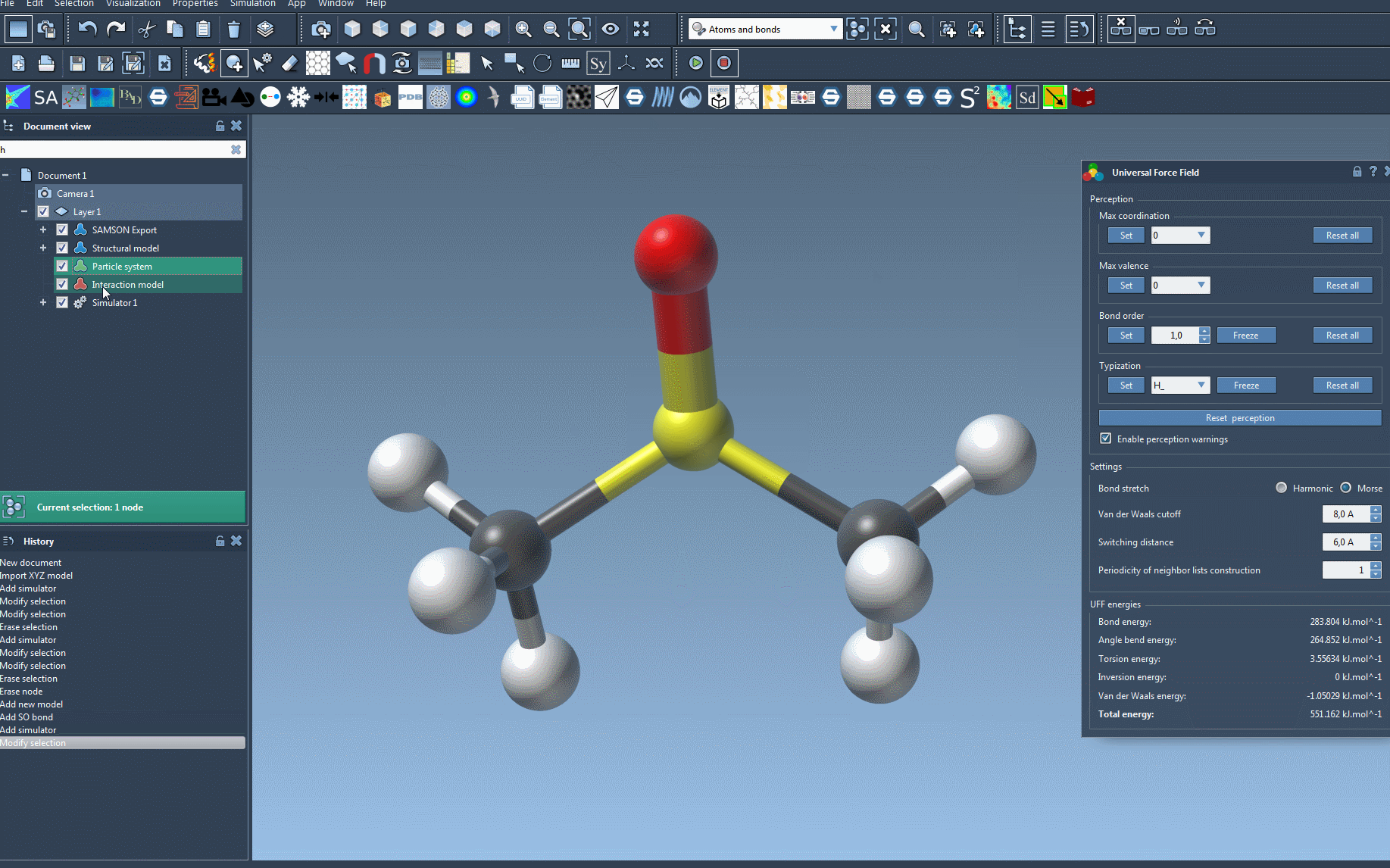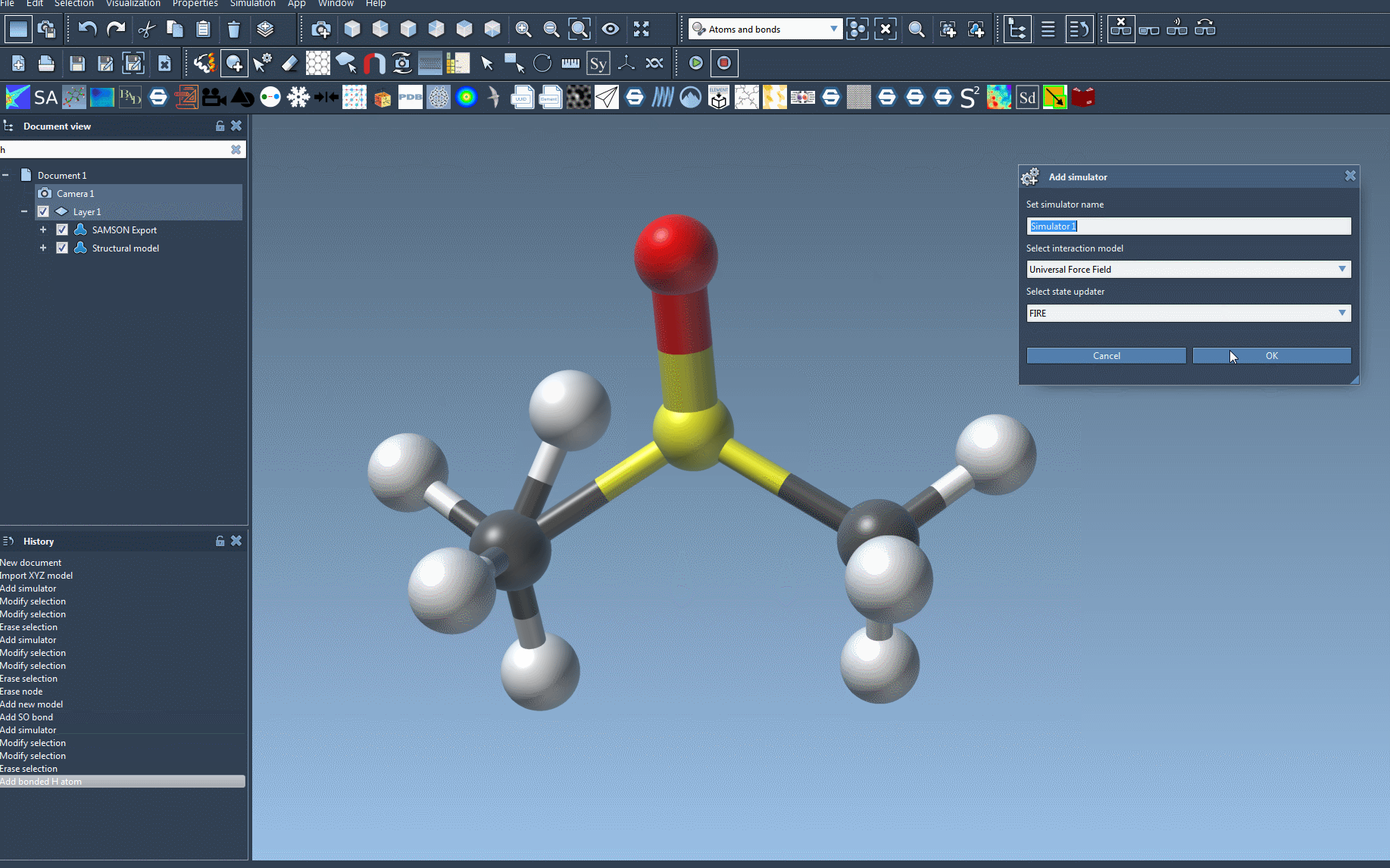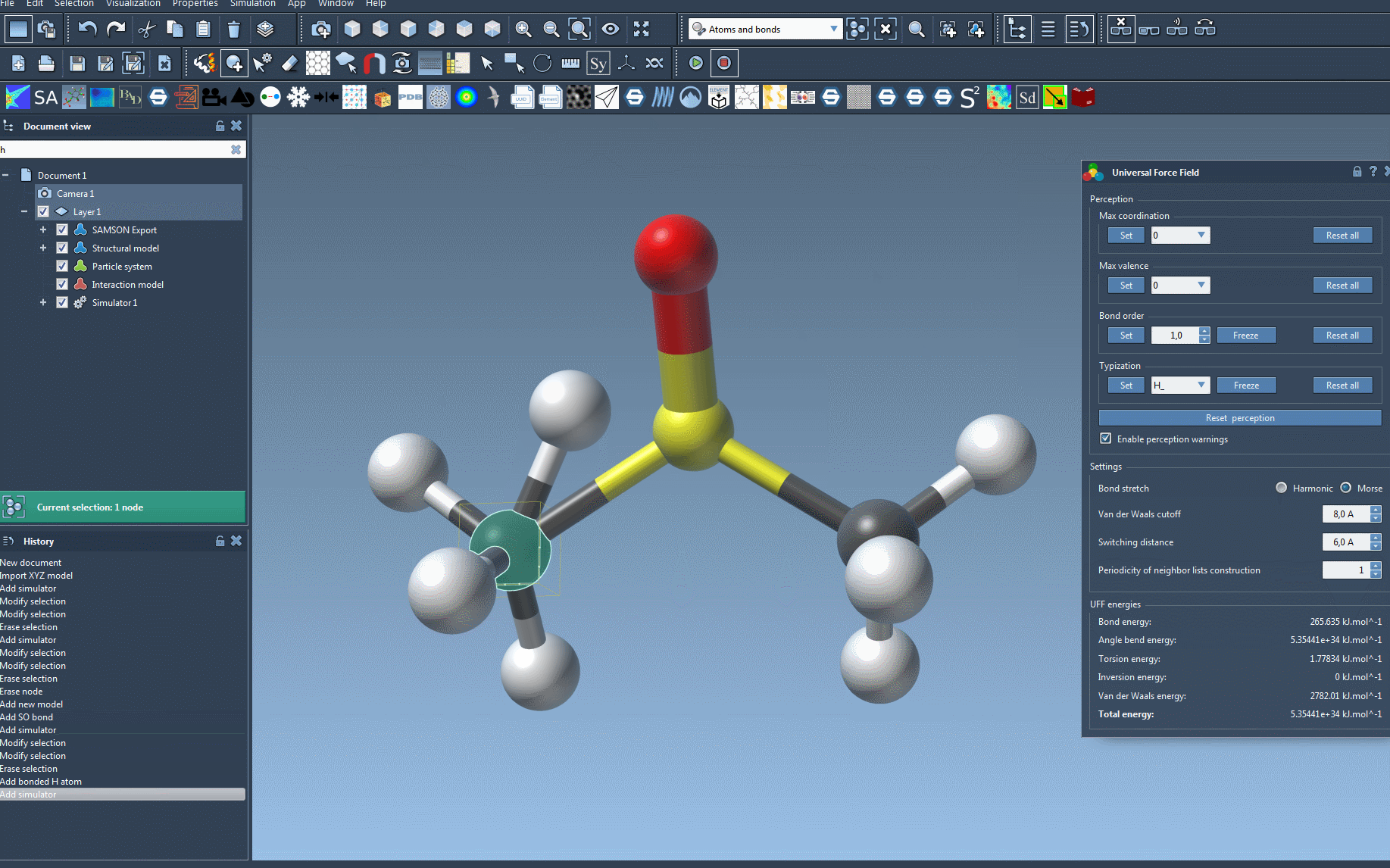
- Add the simulator via Edit > Simulate > Add simulator or use the shortcut:
Ctrl + Shift + M(Windows) orCmd + Shift + M(Mac). - Select Universal Force Field from the list of interaction models.
- Choose a state updater that drives the simulation. For example, you could use the Fast Inertial Relaxation Engine (FIRE) updater.
- Press OK to confirm the setup.
SAMSON will then initialize UFF by automatically computing bonds, bond orders, and atom types if required. This ensures that UFF is ready to simulate the molecular system directly, simplifying what could otherwise be a labor-intensive setup process.
Running the Simulation
Ready to start your simulation? Follow these steps:
- Go to Edit > Simulate > Start to begin the UFF simulation.
During the simulation, the interface displays the energy contributions of different UFF terms. You can even interactively move atoms and observe how the force field dynamically updates the positions of other atoms in response. For additional control, the UFF interface lets you adjust various parameters:
- Bond stretching interactions: Switch between Harmonic and Morse modes depending on the desired accuracy or speed.
- Van der Waals (vdW) cutoff distance: Configure the cutoff distance for vdW interactions.
- Switching distance: Adjust the distance from which the vdW interaction smoothly transitions to zero at the cutoff.
- Periodic neighbor list updates: Modify the frequency at which neighbor lists are rebuilt for vdW interactions, trading off efficiency and precision.
Why Use UFF in SAMSON?
UFF in SAMSON is ideal for users looking for a balance of automation and customization. The automatic molecular structure perception feature reduces mistakes in defining the system and saves time, while advanced users retain the option to customize aspects such as bond orders and atom types. From modeling small molecules to large complexes, this flexibility caters to a wide variety of needs.
For researchers, SAMSON’s UFF implementation strikes a balance between ease of use and complete control. No matter your level of expertise, this tool ensures you can carry out simulations with efficiency and precision, letting you focus on what matters most—your science.
Learn More
If you’re eager to dive deeper into running UFF simulations or accessing detailed references, visit the full tutorial at SAMSON UFF documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON from this link.