





Protein-ligand docking is a critical step in molecular modeling and drug design, but setting up the system correctly can often seem daunting. If you’re short on time or unsure where to start, the AutoDock Vina Extended SAMSON Extension makes it easier than ever to prepare your docking systems efficiently while avoiding common pitfalls.
Here, we’ll focus on a streamlined, yet powerful, part of the process: setting up receptors and ligands for docking, highlighting options for flexible side chains and rotatable bonds. The aim is to save you time while maximizing accuracy.
Why Proper Setup Matters
Setting up a receptor and ligand accurately is not just busywork. Incorrect settings—like missing hydrogens or omitting flexibility where required—can lead to unreliable docking results or unnecessarily long computation times. Fortunately, the AutoDock Vina Extended extension simplifies these tasks.
Setting Up the Receptor
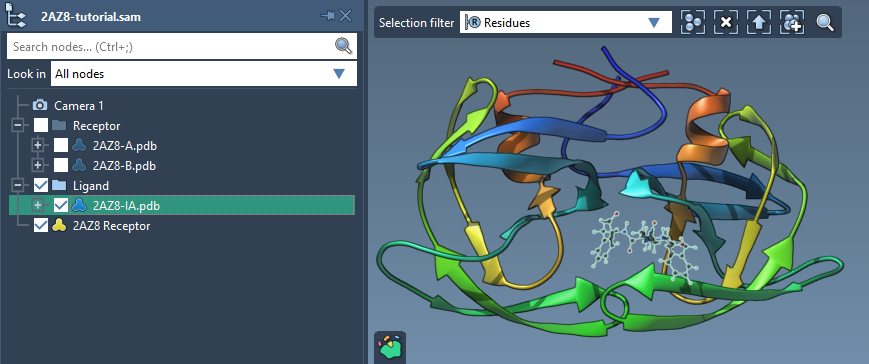
The receptor can be a single model or a receptor library. To assign your receptor, follow these steps:
- Select the receptor (e.g., 2AZ8-A and 2AZ8-B) in the Document view and click Set in the Set receptor part of the interface.
- If needed, use the Select > Biology > Receptors command for auto-selection.
This automatically configures the search domain, which will appear as a yellow box in the Viewport. You can adjust this box manually or use a ligand or specific pocket for reference.
Flexible Side Chains
For docking to flexible receptors, you can enable side chains to rotate. Although this increases docking time, it provides more realistic results. Here’s how to set it up:
- Select residues in the Document view or directly in the Viewport with the Selection filter set to Residues.
- In case of a bound ligand, you can auto-select binding site residues using Select > Biology > Binding sites.
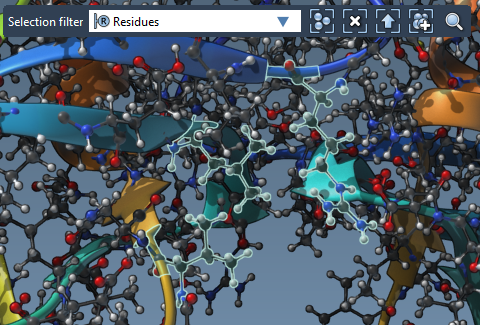
- Click the Set button to assign flexible side chains. These appear as green cylinders in the Viewport, indicating rotatable bonds. Click the bonds to toggle rotational flexibility (green for flexible, red for fixed).
Preparing the Ligand
AutoDock Vina Extended offers options for single ligands or libraries. For a single ligand, select the molecule in the Document view, check the Single ligand option, and click Set.
Adjusting Rotatable Bonds
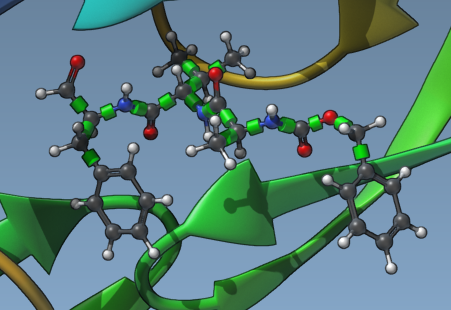
Ligand flexibility is critical for docking accuracy. Once the ligand is set, you’ll see rotatable bonds as green cylinders. You can:
- Toggle specific bonds between flexible (green) and locked (red) by clicking them in the Viewport.
- Use the Locked bonds settings panel to specify and enforce global rules for locking bonds by type (e.g., planar bonds or double bonds).
If your library includes many ligands, selecting the Lock specific ligand bonds option ensures consistent settings across all molecules.
Ligand Minimization
If your ligands aren’t already minimized or include 2D structures, enable the Minimize option. Choose a minimization preset and ensure hydrogens are added if missing. This step ensures ligands are in an energetically favorable state for docking while keeping active bonds flexible.

Final Thoughts
Setting up your system thoughtfully can make all the difference in your docking results. With AutoDock Vina Extended’s intuitive interface, quick receptor/side chain configuration, and flexible ligand handling, you can tackle docking projects with confidence.
To dive deeper into receptor and ligand preparation and explore other aspects of AutoDock Vina Extended, consult the full tutorial at this documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at www.samson-connect.net.