





Molecular modeling often requires identifying precise transition paths between states of a system, such as ligand binding and unbinding or conformational shifts in proteins. These paths help researchers understand key mechanisms, but generating accurate paths can be computationally challenging. Enter the Parallel Nudged Elastic Band (P-NEB) app in SAMSON, a tool that significantly simplifies the optimization process while maintaining accuracy.
What Problem Does P-NEB Solve?
Finding minimum energy paths between molecular conformations is vital for predicting reaction mechanisms or transitions. However, optimizing these paths often involves complex methods that demand time, computational resources, or specialized knowledge. P-NEB tackles this pain directly: it efficiently optimizes transition paths by minimizing energy, while maintaining evenly spaced intermediate conformations. The app can also run computations in parallel for faster results—a major advantage for researchers working with large systems.
How Does It Work?
P-NEB applies spring forces between neighboring conformations to ensure even spacing during optimization. This process helps the method traverse smoothly between known conformations (e.g., local energy minima) while minimizing energy at every step. The app also supports powerful features, such as the climbing image approach to find saddle points. Here’s how you can use it effectively:
1. Set Up Requirements
You’ll need the P-NEB Extension and the FIRE optimizer. With them installed, you’re ready to load your input models.
2. Open the Input Model
SAMSON provides sample trajectories to get you started. For example, you can download the Zinc ligand unbinding trajectory for testing or a more complex protein-ligand complex of Lactose permease and Thiodigalactosid. Once downloaded, your goal is to refine these paths for enhanced accuracy.

3. Optimize with P-NEB
Launch the P-NEB app via Home > Apps > All > P-NEB. Once open, the intuitive interface offers several settings to customize your optimization:
- Spring constant: Defines the elasticity between conformations (e.g., 1.00).
- Number of loops: Determines the iterations for path optimization (e.g., 100).
- Interaction model: Select a force field like “Universal Force Field” for calculations.
- Parallel execution: Check this option for faster computations across conformations.
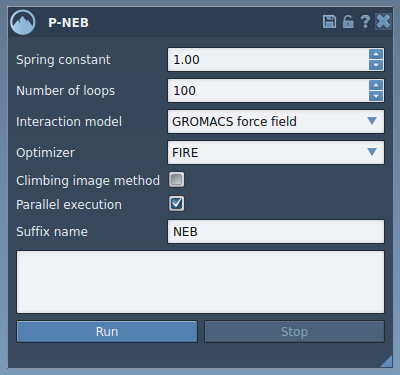
Select your desired path or group of conformations in the Document view, adjust the above settings, and hit Run. During computation, a progress bar will update you on the status:

4. Review the Results
Once finished, P-NEB generates a new optimized path or conformations in the Document view. You can animate these results by double-clicking or inspect details using the Inspector. Additional customization options are available via context menus.
Why P-NEB Stands Out
Applying P-NEB to a path is generally faster than working with individual conformations, but both approaches have their use cases. This adaptability, along with its parallel execution capability, makes P-NEB ideal for researchers who value efficiency without compromising accuracy. Moreover, its user-friendly interface lowers the barrier for molecular modelers of all experience levels.
If you’re ready to streamline your transition path optimization, explore the full documentation for P-NEB at this link.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON from SAMSON Connect.