





When working on molecular modeling, one of the most common challenges encountered is setting up simulations. Whether you’re a beginner or an advanced user, navigating the setup process can feel complex. But with SAMSON’s Universal Force Field (UFF) integration, this process becomes simplified and efficient. Here’s a detailed, step-by-step guide to get you started.
Why Use the Universal Force Field?
The Universal Force Field (UFF) offers full periodic table coverage for molecular mechanics and dynamics simulations, as proposed by Rappe et al. It’s well-suited for a variety of molecular systems. SAMSON’s implementation automates many steps, from perceiving a molecular structure to computing bonds, bond orders, and atom types, enabling direct application of UFF on your model.
Steps to Set Up UFF in SAMSON
Here is a simple guide to launch a UFF-based simulation:
- Open your molecular system: Make sure you’ve loaded the molecular system you’d like to simulate or work with.
- Add a simulator: You can add a simulator through the menu:
Edit > Simulate > Add simulator. For convenience, you can also leverage a shortcut (Ctrl+Shift+M on Windows or Cmd+Shift+M on macOS). - Select Universal Force Field: In the list of interaction models, choose Universal Force Field (UFF). The interface makes it easy to identify and select.
- Choose a state updater: Specify the state updater for your simulation, such as the Fast Inertial Relaxation Engine (FIRE) for geometry optimization.
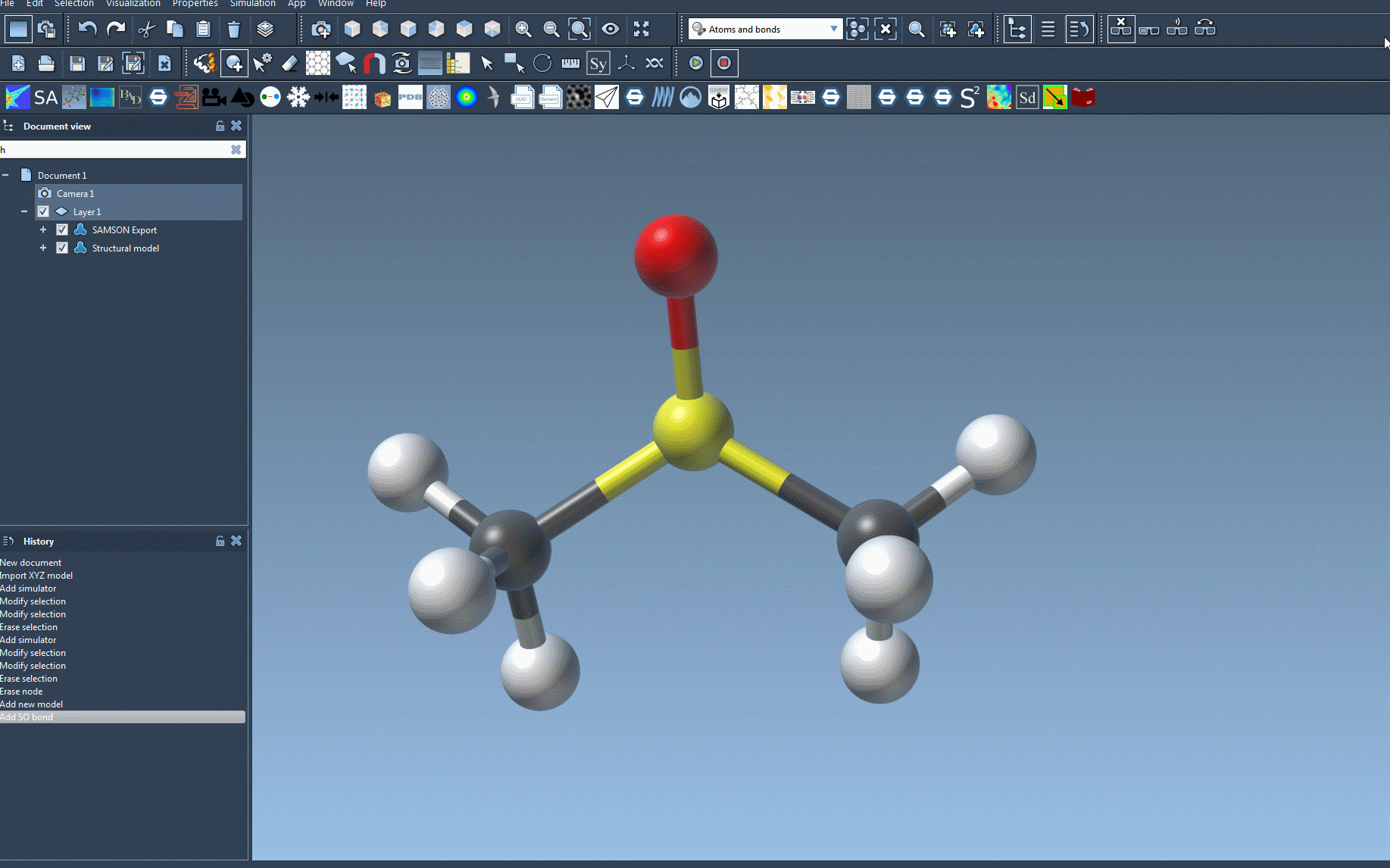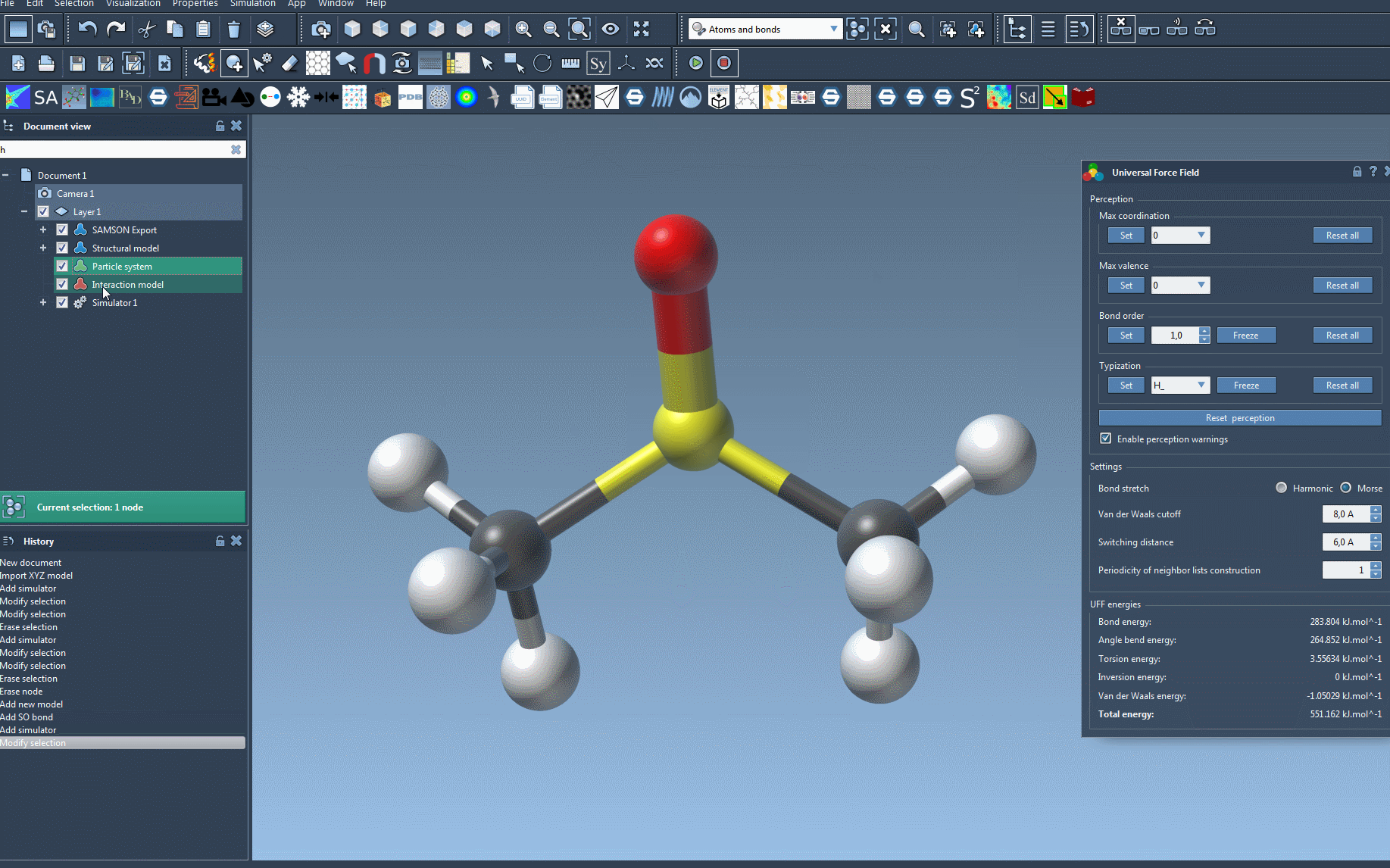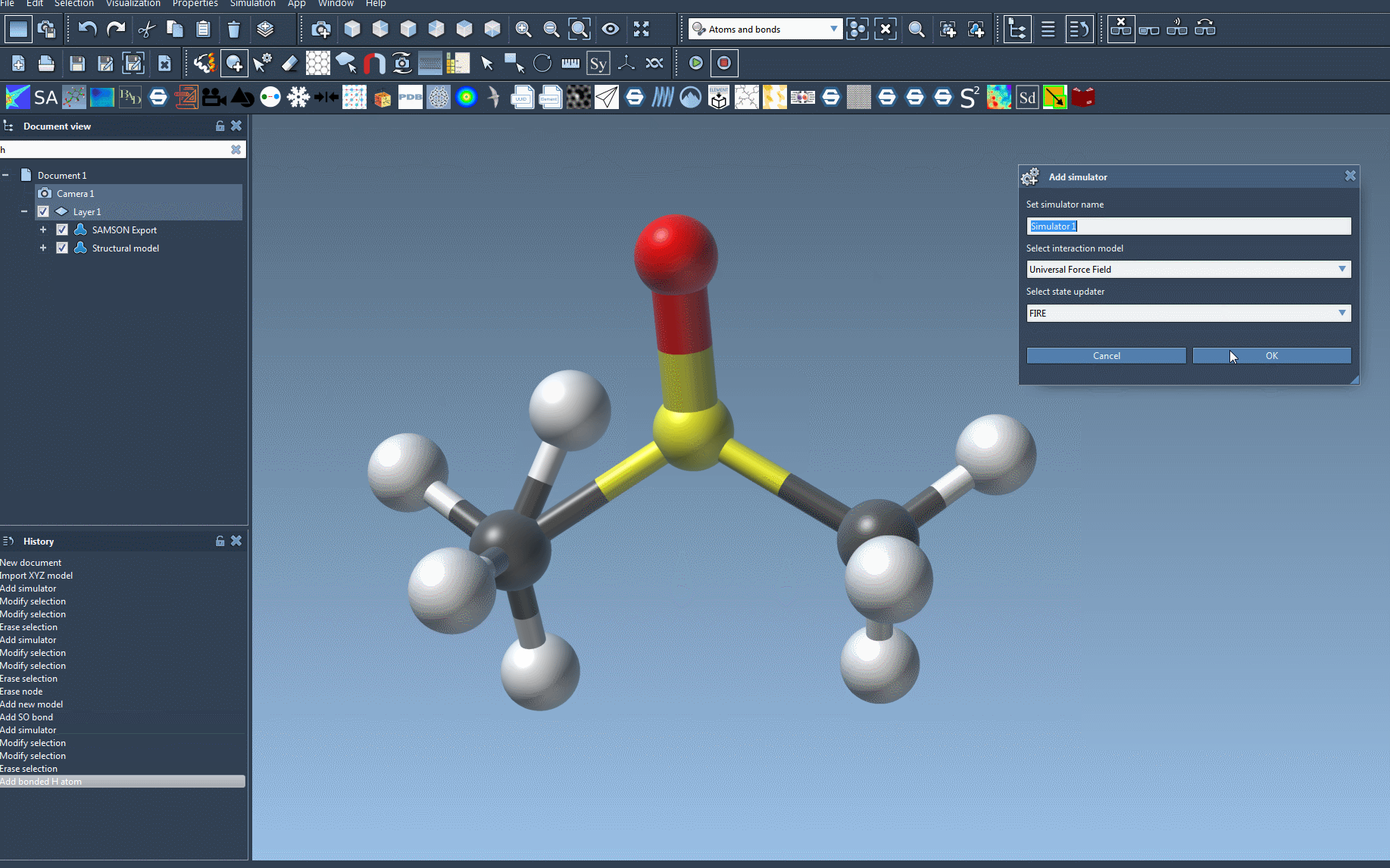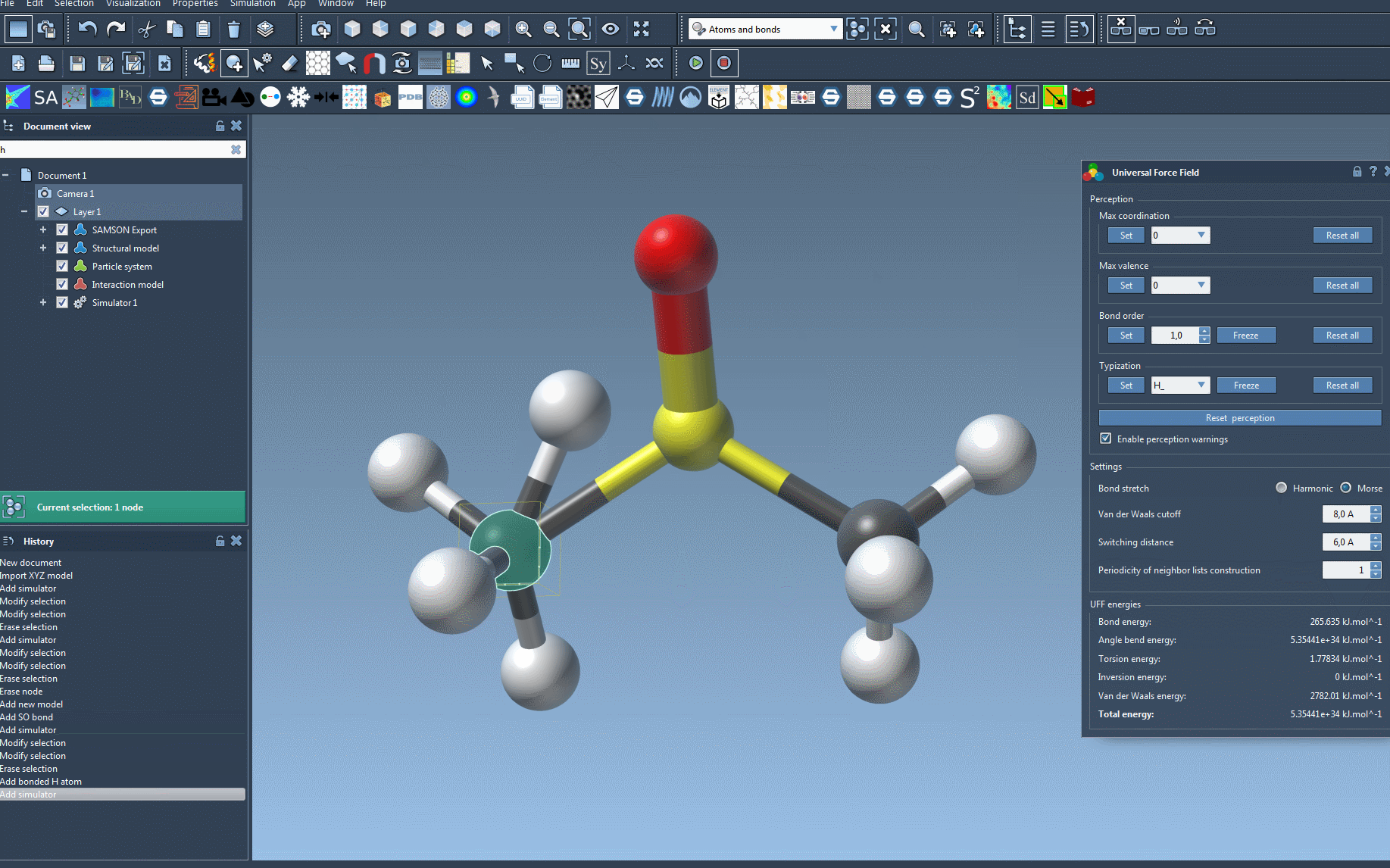
- Finalize setup: Confirm your choices by pressing the OK button. Here, you will now see the UFF setup window.
The UFF setup window further offers the option to rely on existing covalent bonds or allow the program to recompute bonds, bond orders, and atom types automatically. This flexibility accommodates both users with pre-constructed structures and those building from raw data.
Initialization and Potential Challenges
Once initialized, the system computes covalent bonds (if not using existing ones), bond orders, and atom types to prepare your model for simulation. Any detected inconsistencies will result in warnings — an invaluable feature to alert you of potential errors in your model setup.
Upon successful initialization, you’ll find yourself at the UFF Parameters window ready to tweak additional options for your model.
Start the Simulation
Now that the setup is complete, you can begin your simulation:
- Click Edit > Simulate > Start to launch the process.
The UFF interface permits live adjustments and monitoring. For example, you can move atoms interactively and observe the immediate response from other atoms based on computed forces. Additionally, energy terms — including total system energy — are displayed, enabling clear feedback during the run.
Beyond initiating the simulation, you can explore dynamic parameter customization, such as:
- Switching bond stretch interactions between Harmonic and Morse.
- Adjusting the van der Waals cutoff distance and switching distance.
- Modifying neighbor list construction periodicity to optimize computational efficiency.
Conclusion
The Universal Force Field in SAMSON simplifies molecular simulation setup by combining automation with flexibility. From automatic atom perception to bond adjustments, UFF bridges the gap between theoretical models and practical application. The streamlined process leaves you with more time to iterate and experiment on your systems.
For a deeper dive into this topic and other related tools, visit the official UFF tutorial documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Start exploring molecular modeling by downloading SAMSON at samson-connect.net.