





For molecular modelers, visualizing and optimizing transition paths between molecular conformations can often feel like a tedious puzzle. Paths between states are critical to understanding reaction mechanisms or binding/unbinding processes, especially for systems like protein-ligand complexes. However, crafting and perfecting these pathways efficiently is no small challenge. The Parallel Nudged Elastic Band (P-NEB) app in SAMSON offers a targeted solution, facilitating the optimization of transition paths with streamlined computational power.
In this guide, we’ll uncover how to optimize existing molecular paths step-by-step using P-NEB and how this process offers valuable insights into minimum energy pathways. If you’ve ever been stuck trying to refine molecular trajectories between states, this article is for you.
Understanding the P-NEB app
A successful optimization of transition paths relies on balancing precise energy calculations and manageable structure modifications. The P-NEB app in SAMSON uses the climbing image nudged elastic band (NEB) method to handle this process. It works by optimizing intermediate molecular structures along the path, seeking the lowest energy conformation for each point while maintaining uniform spacing. What’s more, the app supports parallel execution, meaning that it evaluates each structure independently in different threads to greatly improve speed.
Getting started with P-NEB
The first step in this process is loading a path into SAMSON. For a quick start, developers provide sample transition pathways. For instance, users can download a sample Zinc ligand unbinding trajectory or a Lactose permease—Thiodigalactosid unbinding path from the Documentation Center:
- An example for preliminary testing: Zinc ligand unbinding trajectory.
- Complex scenarios for advanced analysis: Protein-ligand complex (Lactose permease unbinding).

Once a path is integrated into SAMSON, it’s time to fire up the P-NEB app. Go to Home > Apps > All > P-NEB or simply search for it using the search functionality.
Optimizing molecular paths with P-NEB
Here’s where the P-NEB app truly shines. Selecting an already existing path and clicking the Run button in the P-NEB app starts the optimization process.
With customization options like spring constant, number of loops, and choice of interaction models (e.g., Universal Force Field), users can fine-tune the parameters as per their needs. The app also includes an option for climbing images, which focuses computational resources on identifying saddle points.
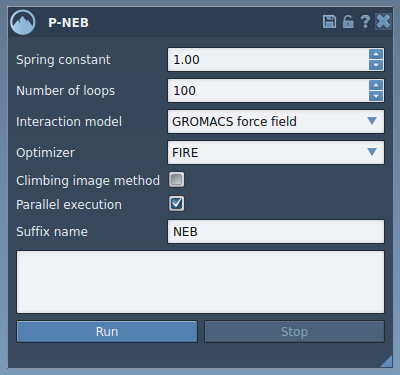
During the execution, users can monitor optimization progress directly in the status bar:

Once the computation completes, a new refined path is seamlessly added to the Document view, ready for review and further analysis. This optimized pathway consistently reflects the lowest energy configurations along the transition route.
Pro tips for molecular modelers
Efficiency tip: If your input model consists only of conformations (and not a path), consider converting these conformations into a single trajectory. Use the Create path from conformations feature under Conformation > Create path from conformations. This shortcut leverages the speed and streamlined calculations of P-NEB on paths rather than sets of conformations.
Experiment strategy: While the default settings work well, try tweaking the spring constant or enabling the climbing image method to observe how it affects the results.
Learn more
Curious to dive deeper? Visit the full documentation on the SAMSON website for a complete guide to optimizing transition paths with P-NEB: Full P-NEB Tutorial.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at SAMSON Connect.