





Setting up interaction models for molecular simulations can often feel like a daunting task, especially when juggling atom types, bond parameters, and interaction specifics. If you’ve ever felt overwhelmed by the process, this post will walk you through the straightforward steps of setting up the Universal Force Field (UFF) model in SAMSON, an integrative platform for molecular design.
Why Universal Force Field (UFF)?
The Universal Force Field (UFF) is a versatile and well-known choice for molecular mechanics and molecular dynamics simulations. Proposed by Rappe et al., UFF features a comprehensive parameter set covering the entire periodic table, making it highly suitable for diverse molecular systems. SAMSON simplifies the application of UFF by introducing an automatic workflow to perceive the molecular structure, compute bond orders, and assign atom types. This means minimizing manual adjustments and setting parameters quickly without compromising accuracy.
How to Set up UFF in SAMSON
The UFF setup process in SAMSON is user-friendly and designed to optimize productivity. Below are the steps to get started:
- Open or load a document containing the molecular system you want to simulate.
- Add a simulation environment by navigating to Edit > Simulate > Add simulator. For quick access, you can use the shortcut
Ctrl+Shift+M(orCmd+Shift+Mon macOS). - Select Universal Force Field from the list of interaction models.
- Choose a state updater (e.g., Fast Inertial Relaxation Engine (FIRE)) to optimize the simulation behavior. Not sure where to start? Default settings are great for first-time users.
- Confirm your selection by pressing the OK button.
You will now see the UFF setup window. In this window:
- Choose whether to Use existing bonds. If unchecked, the system will automatically compute new bonds based on atom types and positions.
- Press the OK button to proceed and start initializing the system.
What Happens Next?
Once the UFF is initialized, the system computes the necessary molecular parameters:
- Calculates covalent bonds (if Use existing bonds was not selected earlier).
- Determines bond orders.
- Assigns UFF atom types based on molecular structure.
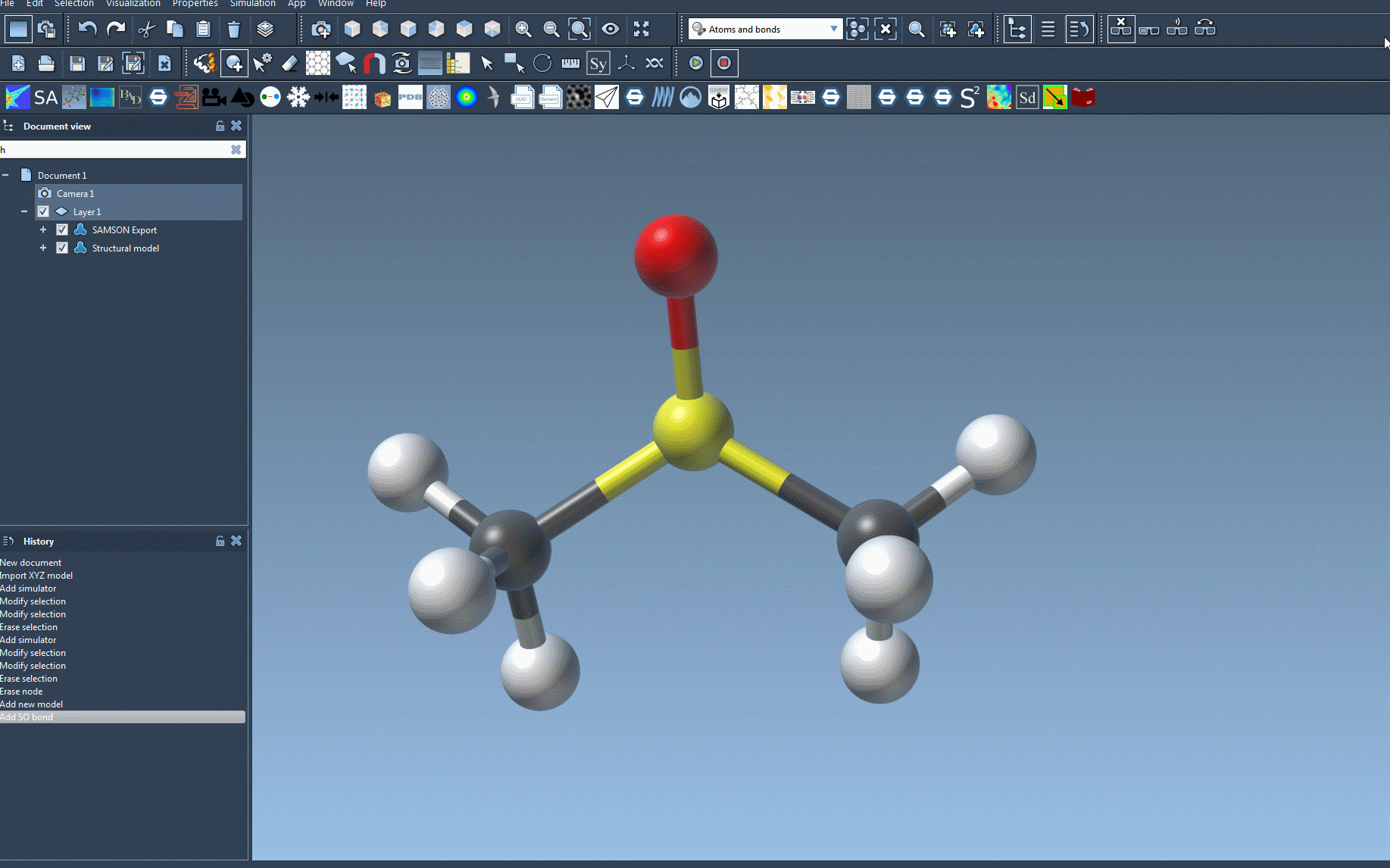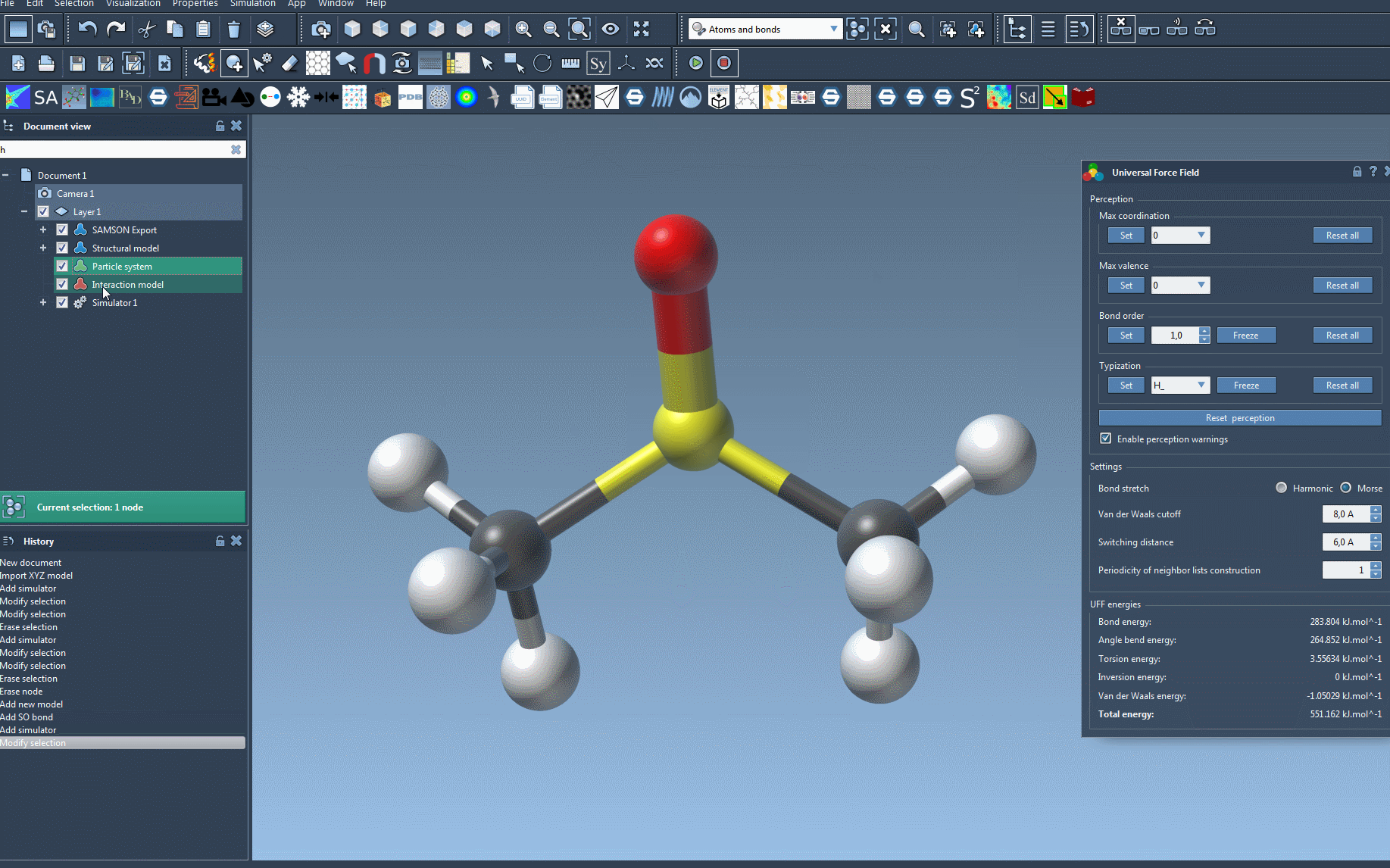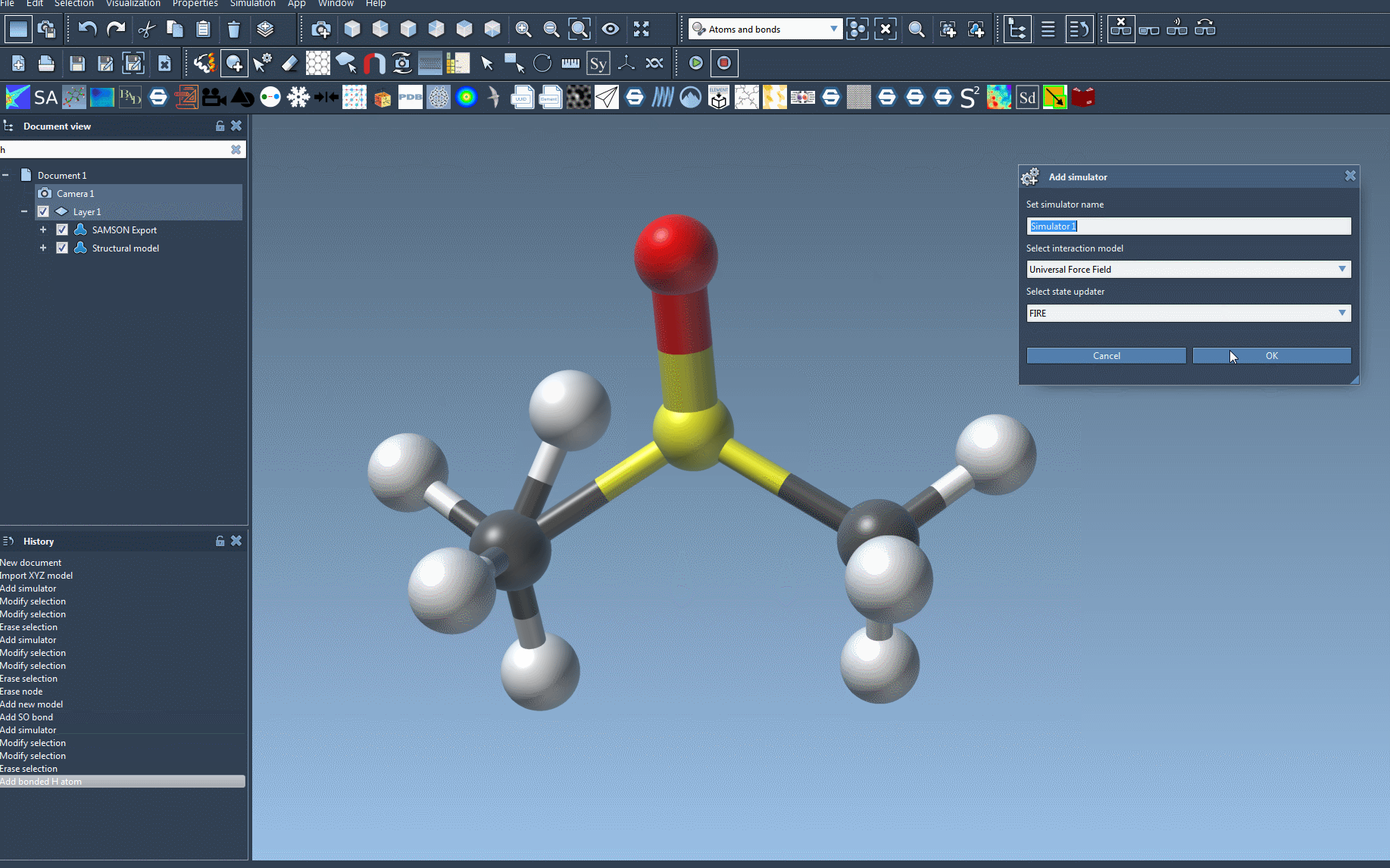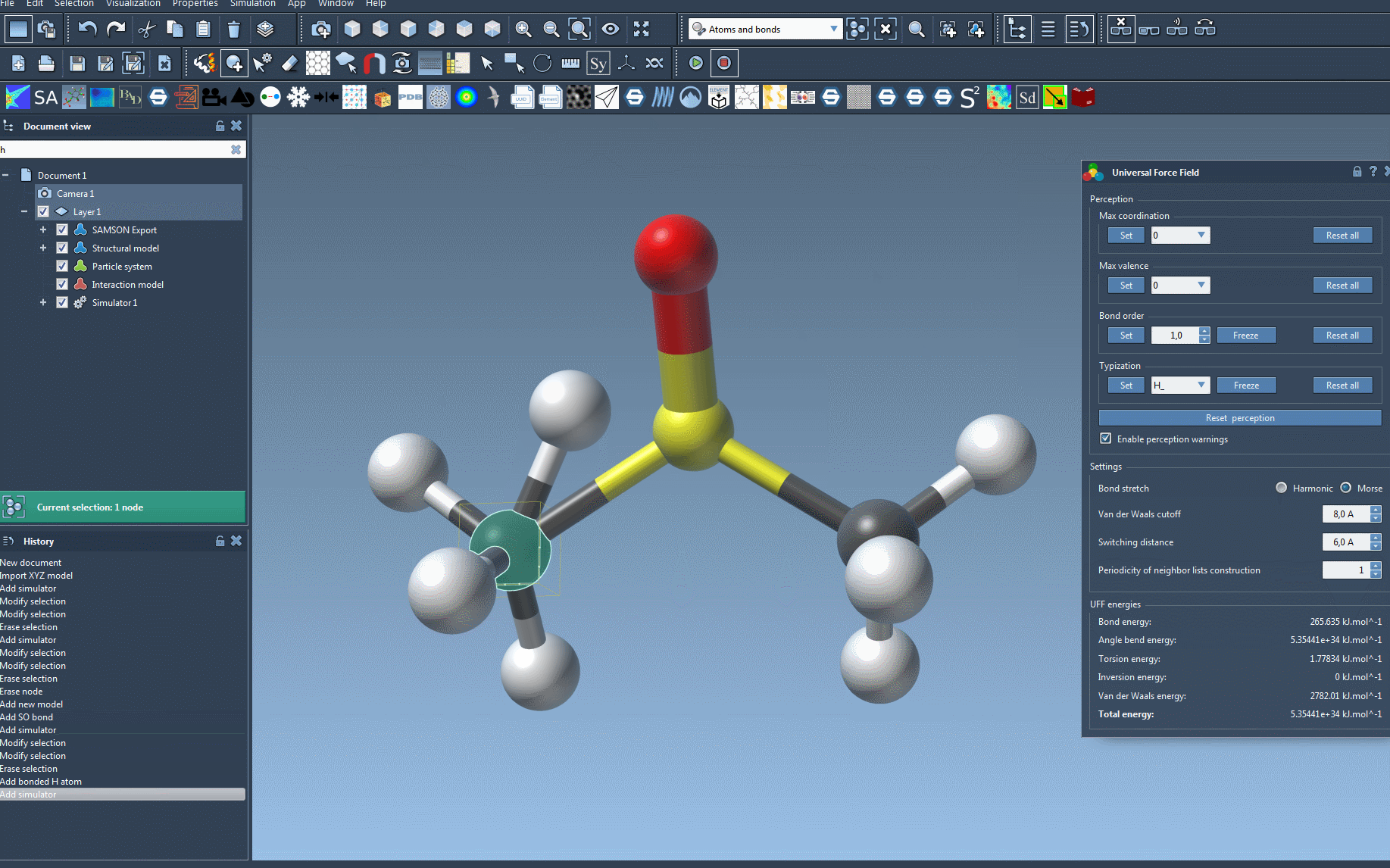
The UFF parameter window will then appear, showing various configuration options for further customization and visualization of interaction terms, such as:
- Switching between Harmonic and Morse bond stretch interactions.
- Adjusting the Van der Waals cutoff distance and the associated switching distance to fine-tune nonbonded interactions.
- Controlling periodicity for neighbor-list updates, affecting computational performance.
With UFF properly set up, you’re ready to simulate. You can start the simulation by selecting Edit > Simulate > Start. During the simulation, SAMSON dynamically updates the system’s energy values and allows you to interactively move atoms to see how neighboring atoms respond.
Visualize the Process with Step-by-step Guidance
Visual learners will appreciate SAMSON’s documentation that includes clear animations demonstrating the UFF setup and molecular simulation in action:
Keep Exploring
This introduction covers the essentials of setting up UFF in SAMSON. For further details on interacting with parameters, advanced customization options for atom typization, or adding and deleting atoms during simulations, consult the complete step-by-step guide in the UFF Documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get started today by visiting SAMSON Connect.