





Setting up molecular simulations can often be a daunting task, especially for newcomers who are exploring force field methods. For molecular modelers working with the Universal Force Field (UFF), the process is vital yet complex. SAMSON, the integrative molecular design platform, offers a systematic and user-friendly way to get started. If you’re looking to efficiently set up UFF in your molecular modeling workflow, this overview will help you get started.
Why the Universal Force Field (UFF)?
The UFF is a comprehensive general-purpose force field, capable of handling the entire periodic table for molecular mechanics and dynamics simulations. It’s particularly powerful due to its automatic perception capabilities, which identify atomic types, bond orders, and covalent bonds directly from the molecular structure. However, efficiently setting it up to run your desired simulations is the first step in realizing its potential, and this is where SAMSON excels.
A Step-by-Step Guide to UFF Setup
Follow these steps within SAMSON to set up UFF in your molecular modeling project:
- Open a document containing the molecular system you wish to simulate. Make sure your molecule of interest is ready for simulation.
- Add a simulator by navigating to
Edit > Simulate > Add simulator. You can also use the shortcutCtrl + Shift + Mon Windows orCmd + Shift + Mon macOS. - In the list of interaction models, select Universal Force Field.
- Pick a state updater for the simulation. For example, the Fast Inertial Relaxation Engine (FIRE) can be an excellent choice for geometry optimization.
- Click OK to proceed.
Once you complete the initial setup, the UFF configuration window will appear. Here’s what you can do in this window:
- Use existing bonds: Select this to rely on bonds already defined for your molecular system. If left unchecked, UFF will automatically recompute the bonds based on atom types and positions.
- Press OK to finalize the setup.
At this stage, the system initializes for UFF by performing tasks such as bond computation, calculation of bond orders, and atom type assignment. If there are inconsistencies detected, SAMSON will notify you with a warning message giving details.
Interactive Features and Customization
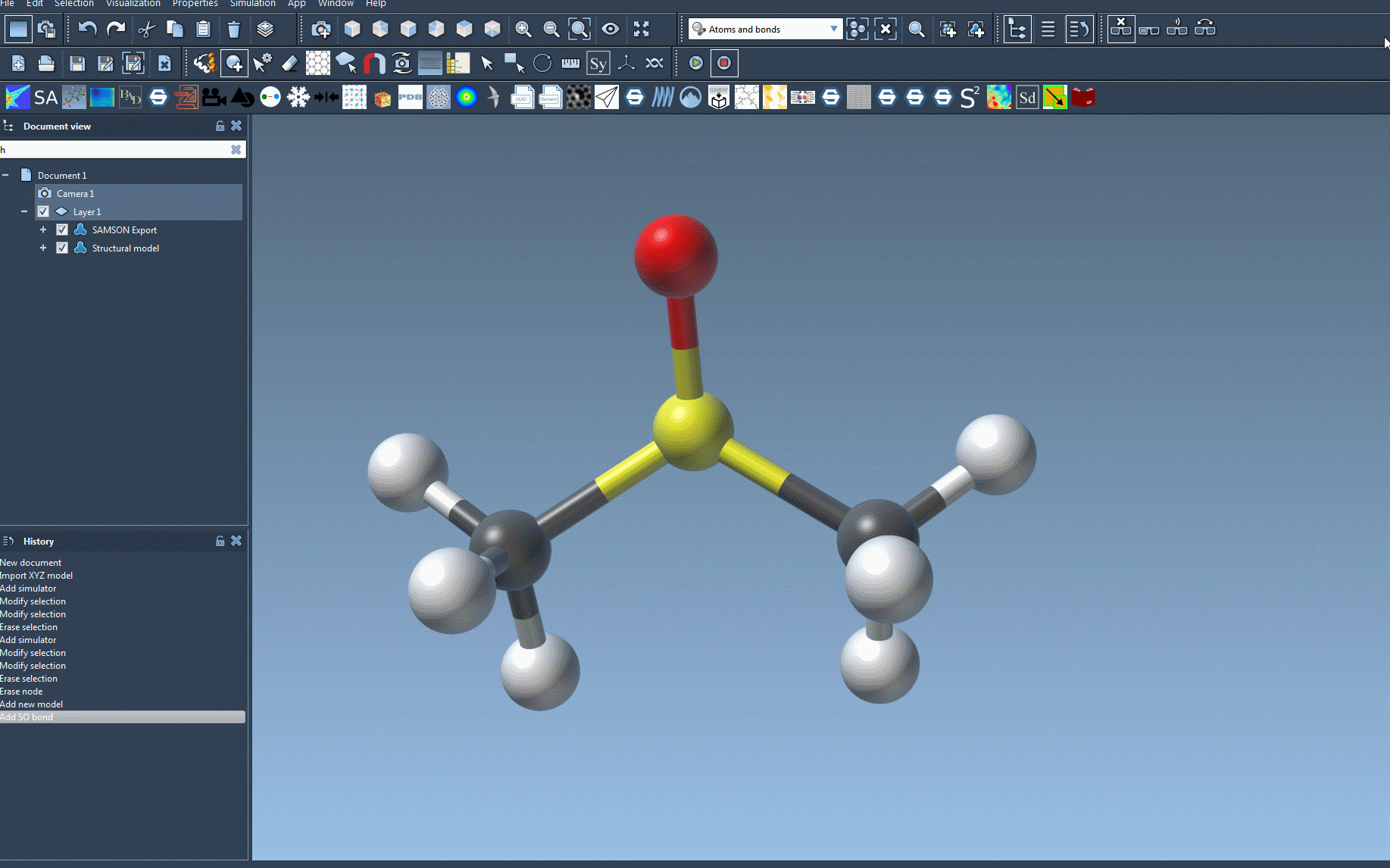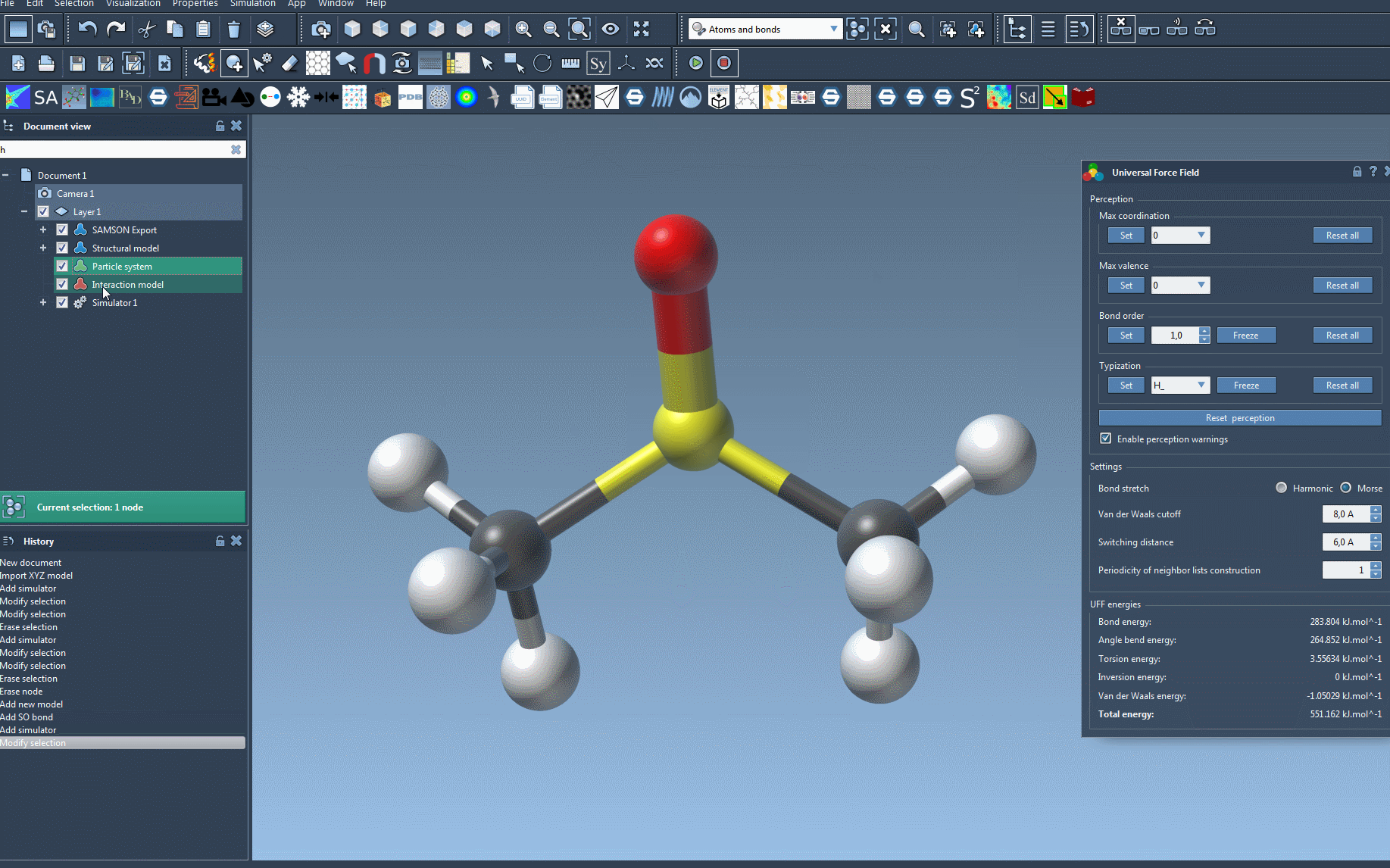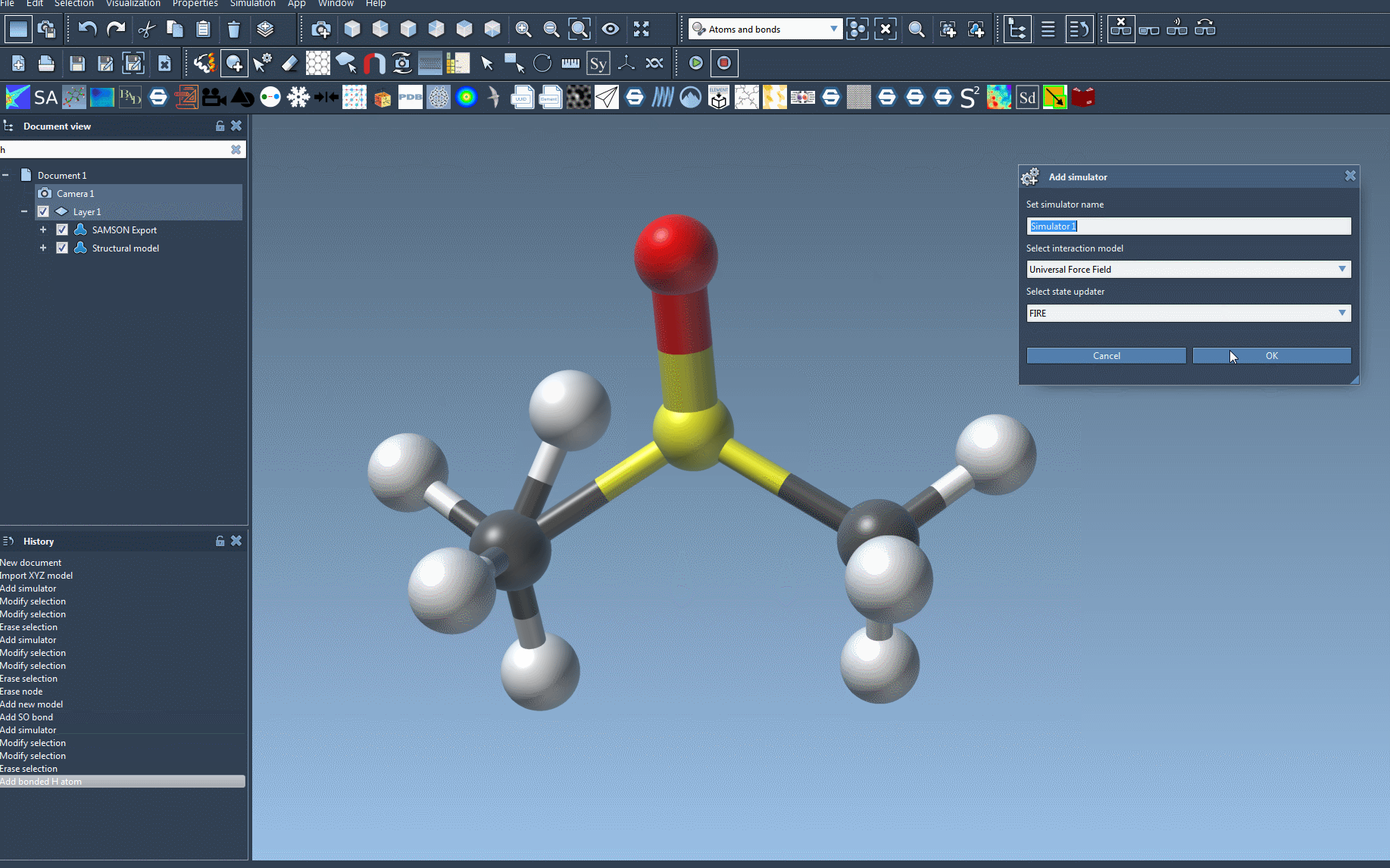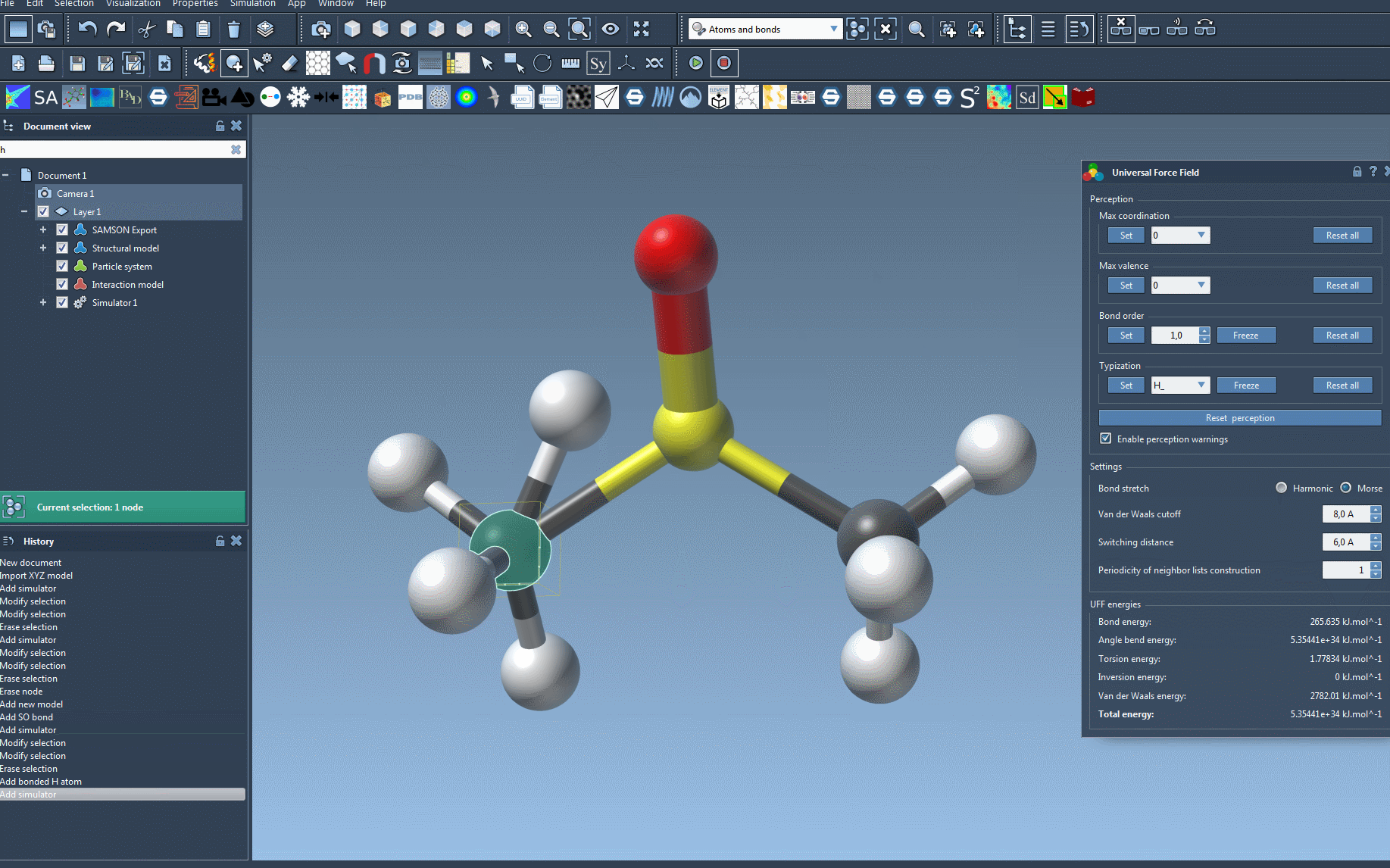
Once your setup is complete, you can use SAMSON’s unique interactivity to explore the behavior of the UFF model. You’ll find the UFF parameter window at your disposal, offering valuable information such as energy terms and the overall simulation energy. The interface also enables users to:
- Adjust bond stretch interaction types (choose between Harmonic and Morse modes).
- Modify van der Waals cutoff distances and switching distances for fine-tuning simulations.
- Control the periodicity of neighbor list construction, influencing its recomputation frequency.
Want to adapt UFF for specific scenarios? Advanced users can dive deeper into typization customization to force bond orders, modify atomic types, or set coordination and valence limits. Remember, these features are powerful but should be used cautiously to avoid erroneous structural setups.
A Note Before You Begin
If it’s your first time using SAMSON’s simulation features, it’s best to start with the default settings and familiarize yourself with the workflow. For further theory, refer to the relevant scientific publications linked in the documentation.
Ready to try it out? To learn more and access the full tutorial, visit the official SAMSON documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at SAMSON Connect.