





For molecular modelers analyzing protein dynamics, modeling realistic transitions between protein conformations can be a challenging and time-consuming task. Whether your research involves conformational analysis, transition state modeling, or setting up umbrella sampling simulations, generating smooth paths between structurally distinct states is often critical. This is where the ARAP Interpolation extension in SAMSON can simplify your workflow.
What is ARAP Interpolation?
The ARAP Interpolator, short for As-Rigid-As-Possible Interpolation, is a powerful tool designed for reconstructing smooth, realistic transition paths between two protein conformations within seconds. The approach ensures the motion is biologically meaningful, making use of geometric models to align and interpolate between conformers. It is an indispensable feature for researchers aiming to better understand structural transitions or prepare reaction coordinates for free energy simulations.
How ARAP Solves an Important Problem for Molecular Modelers
Traditionally, generating continuous paths for molecular dynamics or other simulations often requires manual intervention, scripting, or complex algorithms difficult to implement consistently. The ARAP Interpolation extension in SAMSON automates this process, providing clean, biologically plausible intermediate conformations with minimal user input. This not only saves time but also ensures precise and reproducible results, allowing you to focus more on the analysis rather than the mechanics of path generation.
Using ARAP Interpolation in SAMSON
Step 1: Load and Clean Protein Structures
To begin, fetch your example protein structures directly within the SAMSON interface. For example, load the Protein Data Bank (PDB) identifiers 1DDT and 1MDT for the Diphtheria Toxin, which represent two distinct conformations:
- In the Home > Fetch menu, input
1DDT 1MDTinto the PDB field and click Load.
After loading, you’ll work only with one chain (chain A) by deleting irrelevant chains or molecules like chain B from 1MDT. This ensures a clean dataset for further steps. Finally, click Home > Prepare to automatically remove water, ions, and alternate conformations.
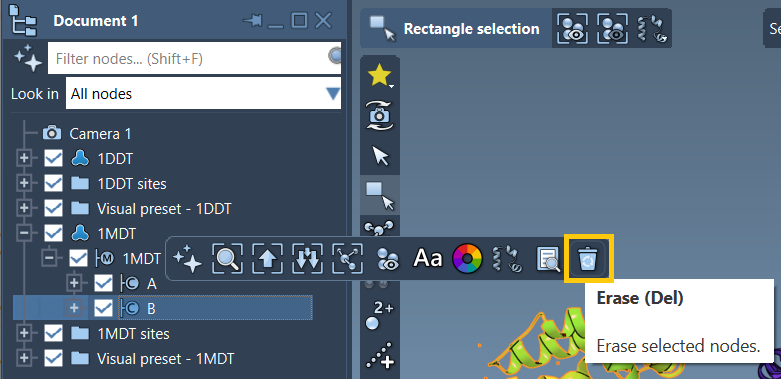
Step 2: Create Conformations
Before running the interpolation, you’ll need to define the start and goal conformations:
- Select
1DDTin the Document view, then go to Edit > Conformation and name it1DDT A. - Repeat the process for
1MDT, naming it1MDT A.
These conformations represent the bookends of your interpolation path.
Step 3: Run the ARAP Interpolator
To generate the transition path:
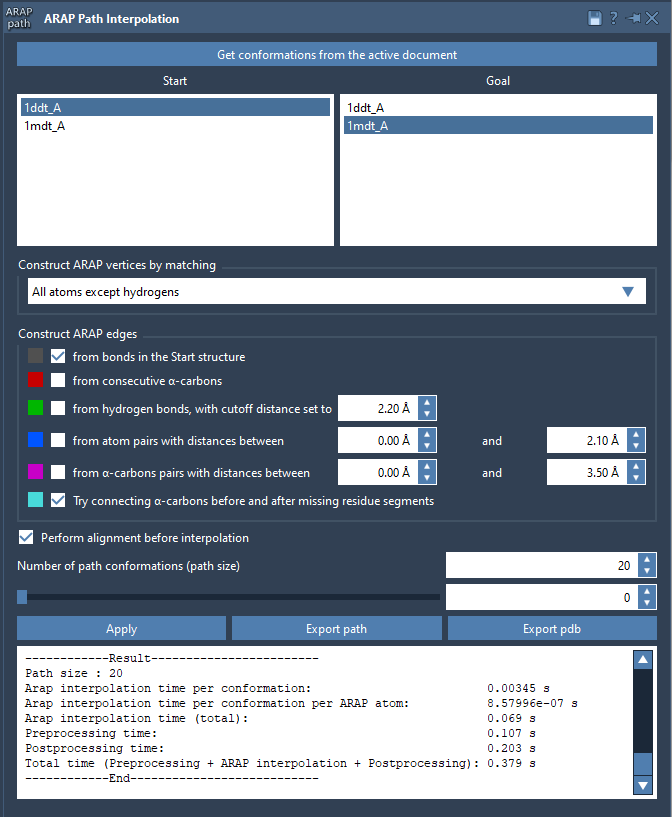
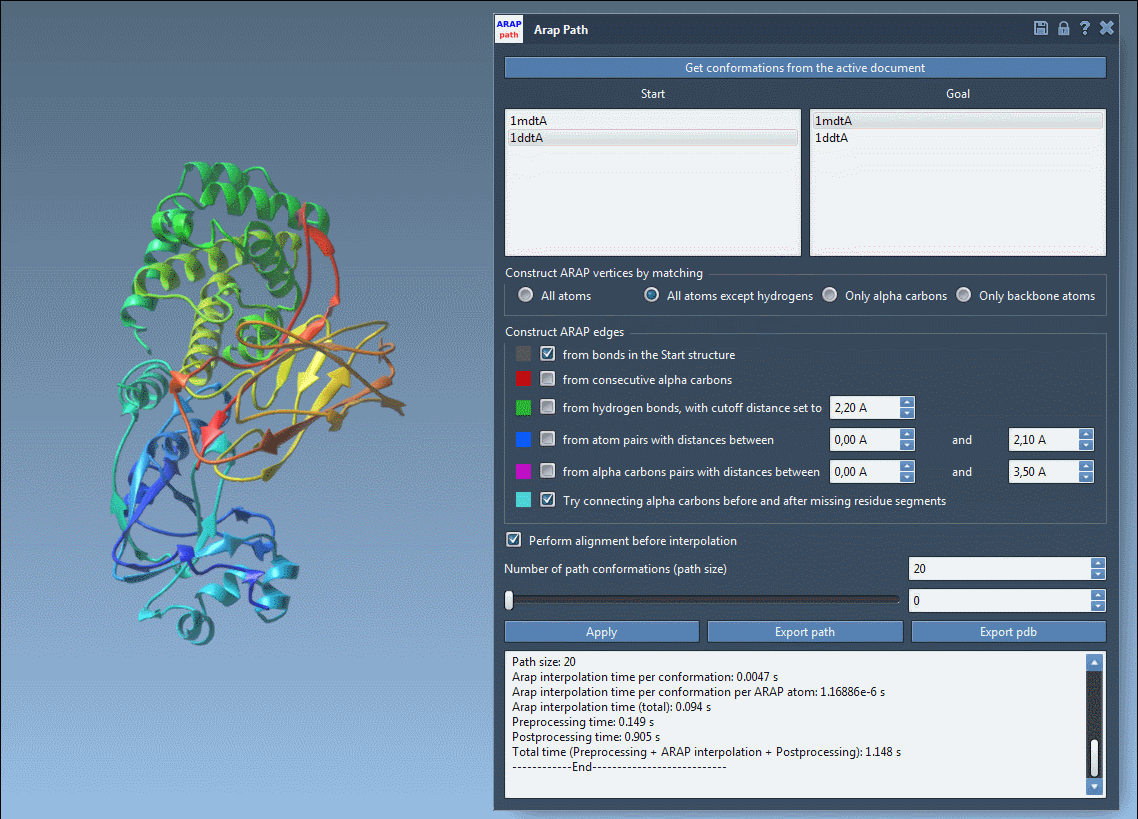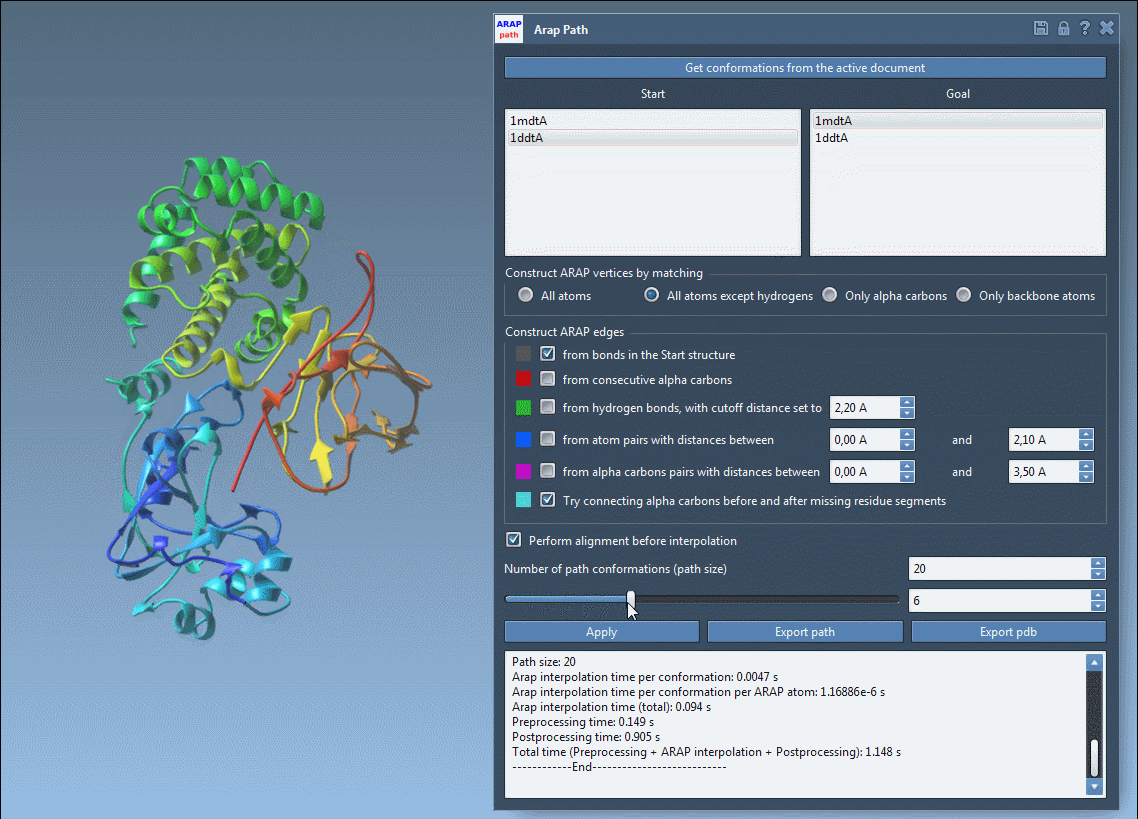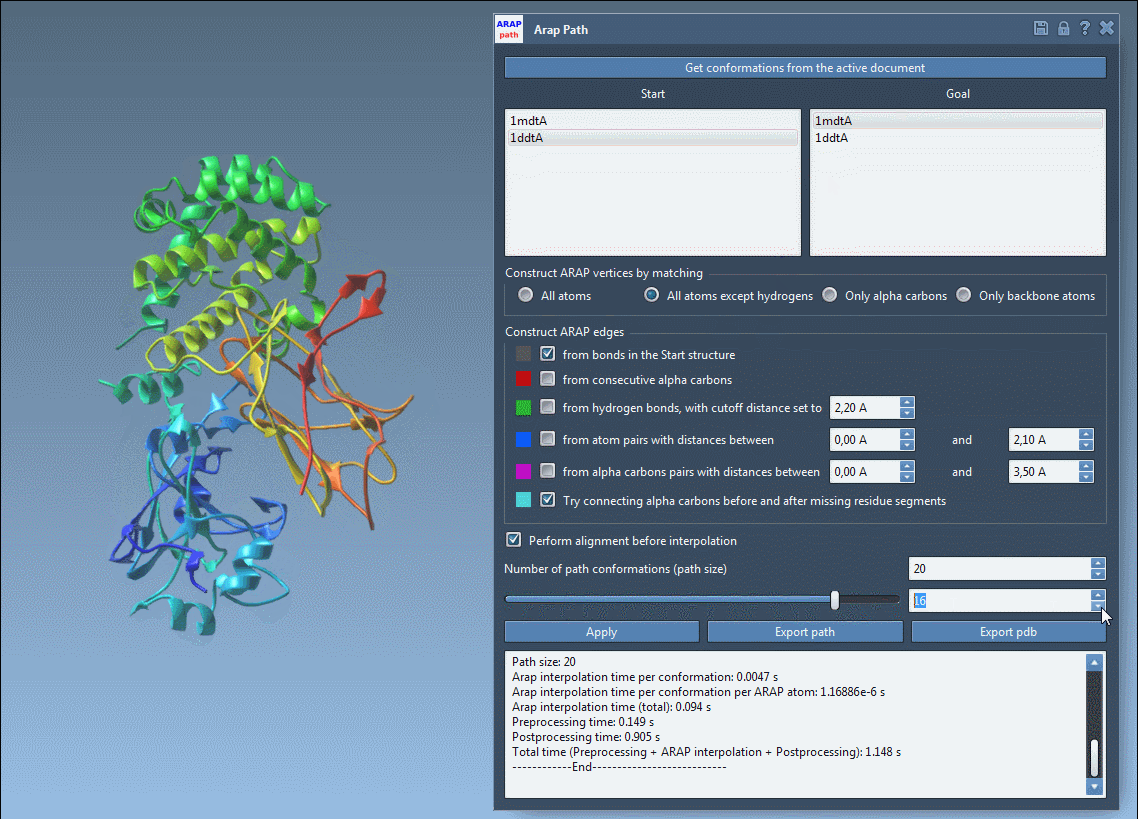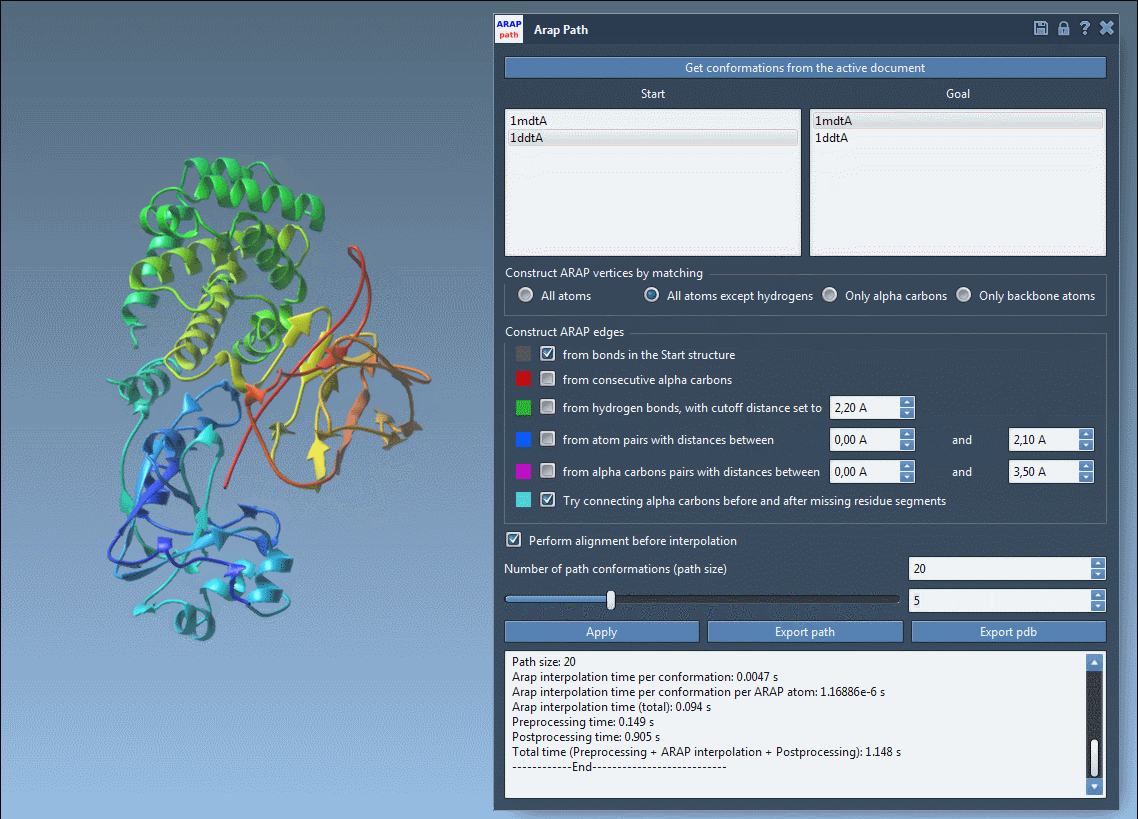
- Open the interpolator app via Home > Apps > Biology > ARAP Path Interpolation.
- Choose your start and goal conformations by clicking Get conformations from the active document and selecting 1DDT A (start) and 1MDT A (goal).
- Set the number of intermediate conformations to generate a detailed trajectory. For example, a count of 20 includes the start and goal conformations.
- Adjust additional settings, such as excluding hydrogens for atom matching or defining how to build ARAP edges to reflect the protein’s backbone and geometric features.
With a single click of the Run button, the app calculates the interpolated path instantly.
Step 4: Analyze and Export
Once the path is computed, you can visualize and analyze the transition. Use the app’s slider to view each conformation along the trajectory and inspect ARAP edges, which can help explain the interpolation’s geometric principles. Finally, export your results as a PDB file or path object for downstream simulations or visualization.
Applications and Further Reading
The ARAP-generated transition paths can serve as input for umbrella sampling, steered molecular dynamics, or nudged elastic band refinements. To dive deeper into these applications, check out the detailed ARAP Interpolation documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. Get started by downloading SAMSON at SAMSON Connect.