





For molecular modelers, one of the most daunting tasks is identifying transition pathways between two conformations of a molecular system. This process is vital for understanding reaction mechanisms, analyzing energy landscapes, or predicting molecular interactions, yet it is often computationally intensive and complex. Thankfully, SAMSON offers an elegant solution with its Parallel Nudged Elastic Band (P-NEB) app, a tool designed to refine transition paths efficiently and effectively.
The P-NEB method is a powerful implementation of the Nudged Elastic Band (NEB) approach, which optimizes a set of intermediate molecular conformations along the transition pathway. By finding the minimum energy paths and maintaining equal spacing between these intermediate states, it ensures a smooth, accurate trajectory between starting and ending conformations. For example, if you have relaxed states of your system and want to find how it transitions between these states, P-NEB can generate precise and meaningful paths.
How Does P-NEB Work?
To apply P-NEB, you’ll first need a path or a set of conformations to define the starting points. These can be generated through linear interpolation or tools available in SAMSON, such as the Ligand Path Finder. P-NEB comes into action when you refine these initial guesses to uncover the optimal pathways.
Here’s how it generally works:
- The app applies spring forces to keep the intermediate conformations spaced evenly.
- It leverages an interaction model such as Universal Force Field (UFF) to compute energies and forces.
- An optimization algorithm, like the Fast Inertial Relaxation Engine (FIRE), drives the refinement of the path.
- An optional “climbing image” strategy can further identify the saddle point by carefully optimizing the energy peak along the path.
Step-by-Step Guide to Using P-NEB
Let’s walk through a typical workflow. Imagine you’re working on a ligand unbinding process, such as the unbinding of Lactose Permease and Thiodigalactosid. Follow these essential steps:
- Load the input model via Home > Download in SAMSON. You can use predefined documents from SAMSON Connect, such as this protein-ligand complex.
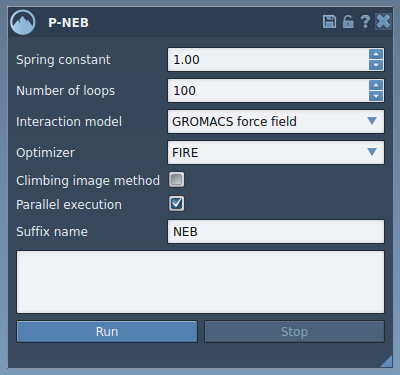
- Launch the P-NEB app: Go to Home > Apps > All > P-NEB. The app interface lets you configure essential parameters like spring constants, number of optimization loops, and the chosen force field.
- Select your target (a path or set of conformations) in the Document view, then hit the Run button in the app to start the P-NEB process.
- Monitor progress in the status bar and examine the final refined path or conformations, which appear as new nodes in the Document view. You can double-click on these nodes to visualize the optimized transition.
Why Choose P-NEB?
The P-NEB app is a smart choice for any molecular modeler looking to improve the quality of transition paths, thanks to its flexibility and ease of use. It’s particularly beneficial for anyone dealing with large systems, as it offers parallel execution (one thread per conformation). This not only saves time but ensures accuracy when working with complex systems.
To optimize workflows further, SAMSON provides the option to convert sets of conformations to a single path via the Document view. This functionality reduces computational load and simplifies visual analysis.
Conclusion
Using the P-NEB tool in SAMSON can help take the guesswork out of determining transition pathways, giving you precise results with minimal effort. If you’re ready to optimize your molecular transitions, follow this guide and explore the full tutorial available here.
SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON at https://www.samson-connect.net.