





When working with protein structures, even small mistakes in backbone conformation can introduce physical strain or mislead simulation outcomes. Before launching into molecular dynamics or interpreting mechanistic insights, it’s helpful to take a moment and check your model’s backbone—especially when working with homology models or low-resolution data.
This is where the Interactive Ramachandran Plot in SAMSON can make a big difference. It offers more than just visualization: it provides an intuitive interface to detect and interactively adjust dihedral angles of residues in your protein model, combining spatial awareness and live 3D updates. Let’s explore how this works.
Residue-by-Residue Visual Inspection
Once your protein is loaded (for instance, using PDB ID 1YRF), open the Ramachandran plot app in Home > Apps > Biology. A 2D plot shows ψ vs. φ angles of each residue, colored by their conformational preference:
- Yellow areas: energetically favorable regions
- White areas: regions to be cautious with
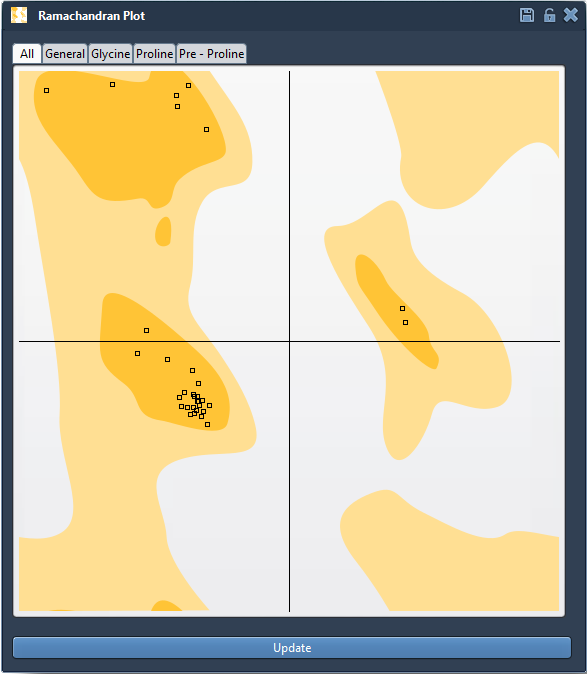
This view supports various residue-specific filters (General, Glycine, Proline, Pre-Proline), making it easier to isolate potentially problematic residues based on stereochemistry.
Live Correction Through Interaction
Seeing a residue in an unfavorable area? You can fix it immediately:
Option 1: Drag Points in the Plot
- Click the residue point
- Drag it toward a favored region on the plot
- The 3D structure updates in real time
This is ideal for quick corrections, and gives tangible feedback as the backbone shape adjusts in the main viewport. If you want to revert, just press CTRL/CMD + Z.
Option 2: Use the Twister Editor
To get a more structural sense of adjustments, select the Twister Editor from the left-hand menu and twist directly in 3D:
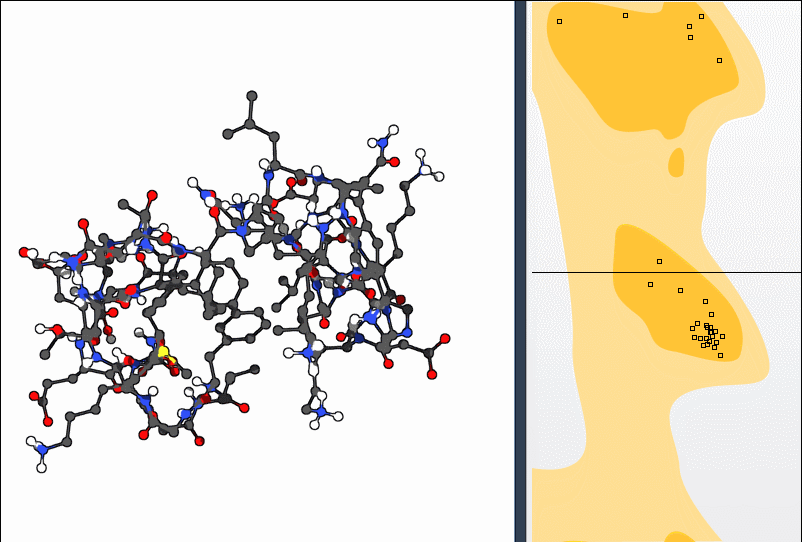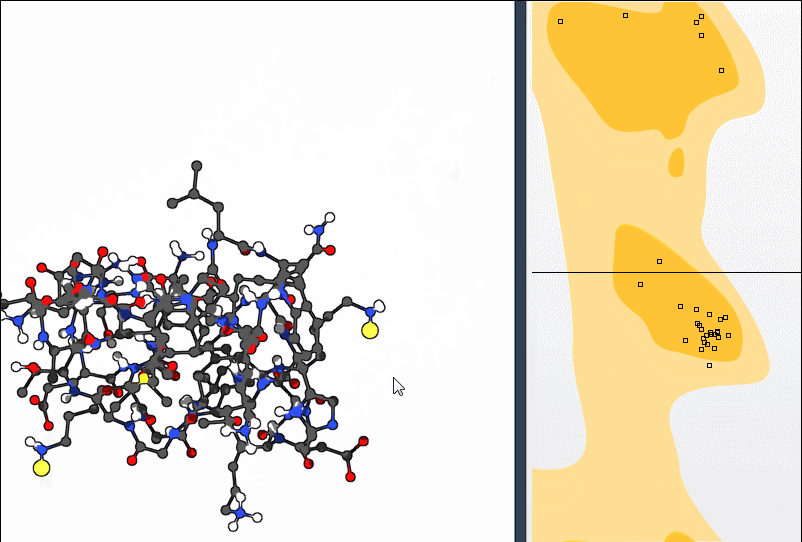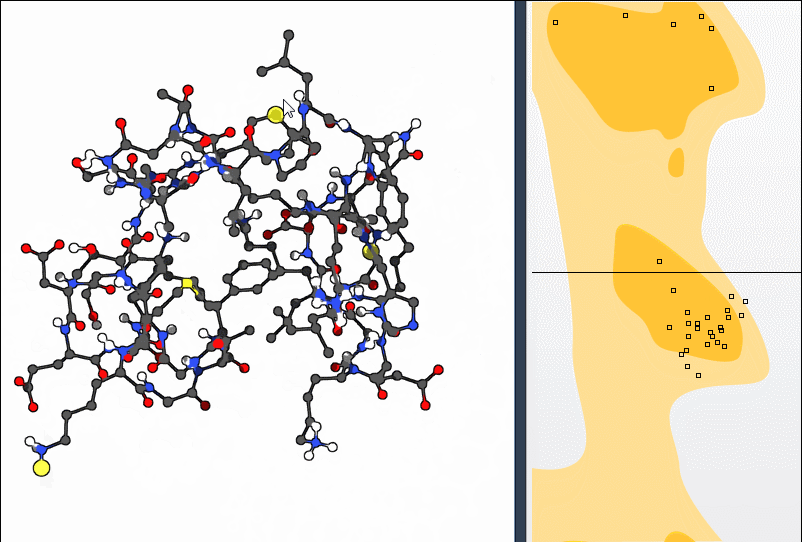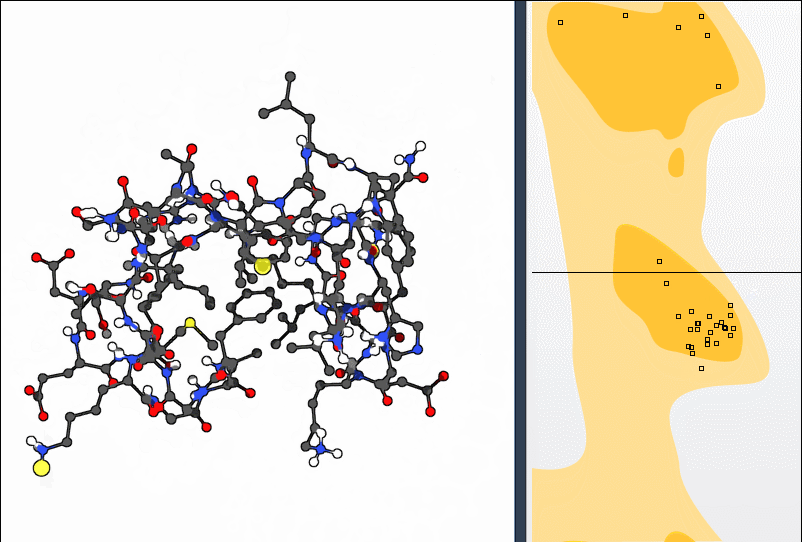
The Ramachandran plot updates as you manipulate the residue, showing how your change affects φ and ψ angles live. This makes it easier to *understand* your action rather than just modify numerically.
When to Use This?
Interactive refinement can help in:
- Fixing unusual conformations in homology models
- Preparing models for simulation by removing high-strain areas
- Validating predicted structures before interpreting binding sites
- Teaching conformational limitations of amino acids
A small investment of time tweaking incorrect dihedrals can prevent issues in downstream analyses like energy minimization, dynamics, or docking.
Want to Learn More?
For a step-by-step guide, check the full documentation here: Interactive Ramachandran Plot Tutorial.
SAMSON and all SAMSON Extensions are free for non-commercial use. To get started, download SAMSON at samson-connect.net.